Exhibit 99.2

December 11, 2025 New York City

Forward-Looking Statements This presentation includes forward-looking
statements that are subject to substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. All statements other than statements of historical facts contained in
this presentation, including statements regarding our future results of operations and financial position, business strategy, potential uses of cash and capital allocation, research and development plans, profitability, the anticipated
timing, costs, design, conduct and results of our ongoing and planned preclinical studies and clinical trials for our products and product candidates, and any commercial potential of our products and product candidates are forward-looking
statements. These forward-looking statements are based upon the current expectations and beliefs of our management as of the date of this presentation and are subject to certain risks and uncertainties that could cause actual results to
differ materially from those described in the forward-looking statements. Although we believe that our plans, intentions, expectations and strategies as reflected in or suggested by those forward-looking statements are reasonable, we can give
no assurance that the plans, intentions, expectations or strategies will be attained or achieved. Furthermore, actual results may differ materially from those described in the forward-looking statements. These forward-looking statements may
be affected by a number of risks, uncertainties and assumptions, including, but not limited to, those risks set forth in the sections captioned “Risk Factors” and “Forward-Looking Statements” of our filings with the U.S. Securities and
Exchange Commission, available at www.sec.gov and investor.roivant.com. We operate in a very competitive and rapidly changing environment in which new risks emerge from time to time. These forward-looking statements are based upon the current
expectations and beliefs of our management as of the date of this presentation, and are subject to certain risks and uncertainties that could cause actual results to differ materially from those described in the forward-looking statements.
Except as required by applicable law, we assume no obligation to update publicly any forward-looking statements, whether as a result of new information, future events or otherwise. This presentation includes data for each of batoclimab,
IMVT-1402, brepocitinib, and mosliciguat as compared to certain other potential competitor products generated from separate, independent studies and that do not come from head-to-head analyses. Differences exist between study or trial designs
and subject characteristics and caution should be exercised when comparing data across studies. Data regarding other products is based on publicly available information. Non-GAAP Financial Information This presentation includes certain
financial measures that were not prepared in accordance with U.S. generally accepted accounting principles (GAAP). Additional information regarding non-GAAP financial measures can be found on slides 166-167. Any non-GAAP financial measures
presented are not, and should not be viewed as, substitutes for financial measures required by U.S. GAAP, have no standardized meaning prescribed by U.S. GAAP and may not be comparable to the calculation of similar measures of other
companies. Disclaimer This presentation is intended for the investor community only; it is not intended to promote the product candidates referenced herein or otherwise influence healthcare prescribing decisions. 2

Today’s Agenda 8:00 – 8:15 Introduction 8:15 – 9:00 Brepocitinib 9:00 –
9:15 Q&A 9:15 – 9:25 Break 9:25 – 10:20 IMVT-1402 10:20 – 10:30 Q&A 10:30 – 10:50 Mosliciguat 10:50 - 11:00 Q&A 11:00 – 11:10 Break 11:10 – 11:25 LNP Litigation 11:25 – 11:30 Financial Outlook 11:30 –
11:40 Closing Remarks 11:40 – 12:00 Q&A 12:00 Lunch For investor audiences only 3

Today’s Speakers Matt Gline CEO, Roivant Ben Zimmer CEO, Priovant Drew
Fromkin CEO, Pulmovant Richard Pulik CFO, Roivant Frank Torti President & Vant Chair, Roivant 4 CEO, Immunovant President, Roivant Eric Venker Lindsay Androski Special Counsel, Genevant CEO, Arbutus

Matt Gline CEO, Roivant 5 Introduction
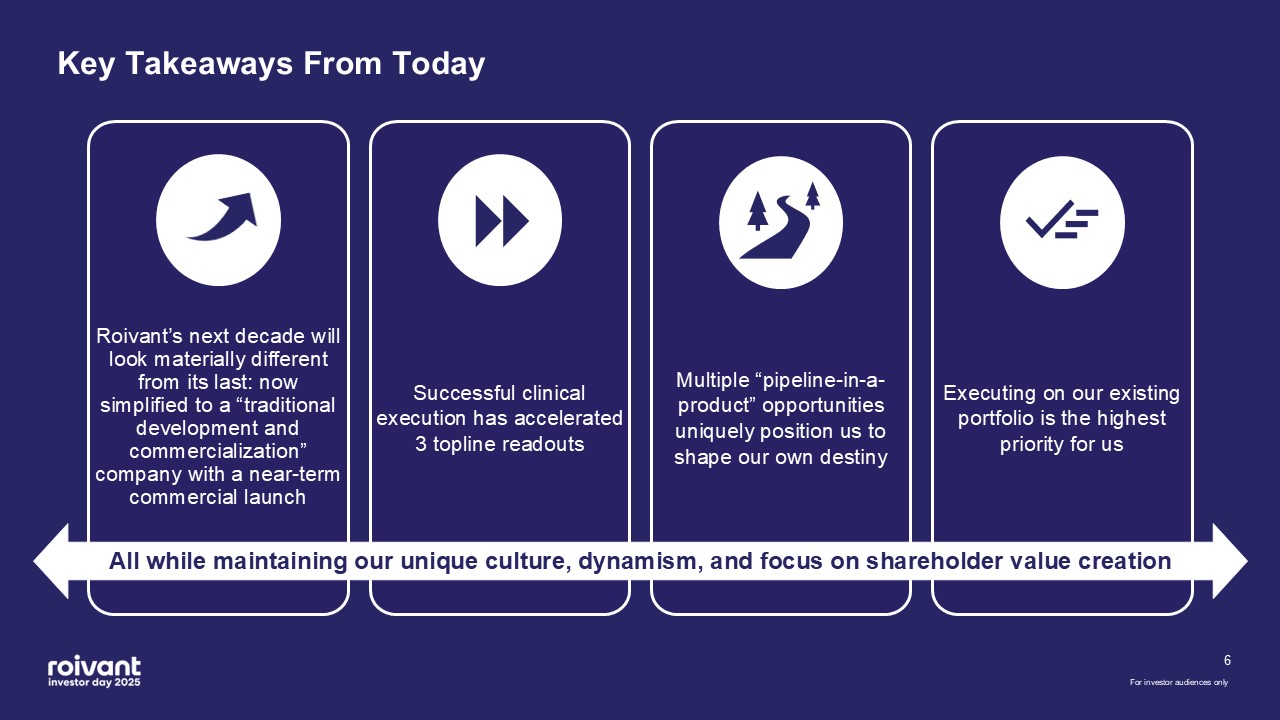
6 Key Takeaways From Today Roivant’s next decade will look materially
different from its last: now simplified to a “traditional development and commercialization” company with a near-term commercial launch Executing on our existing portfolio is the highest priority for us Multiple “pipeline-in-a-product”
opportunities uniquely position us to shape our own destiny Successful clinical execution has accelerated 3 topline readouts For investor audiences only All while maintaining our unique culture, dynamism, and focus on shareholder value
creation
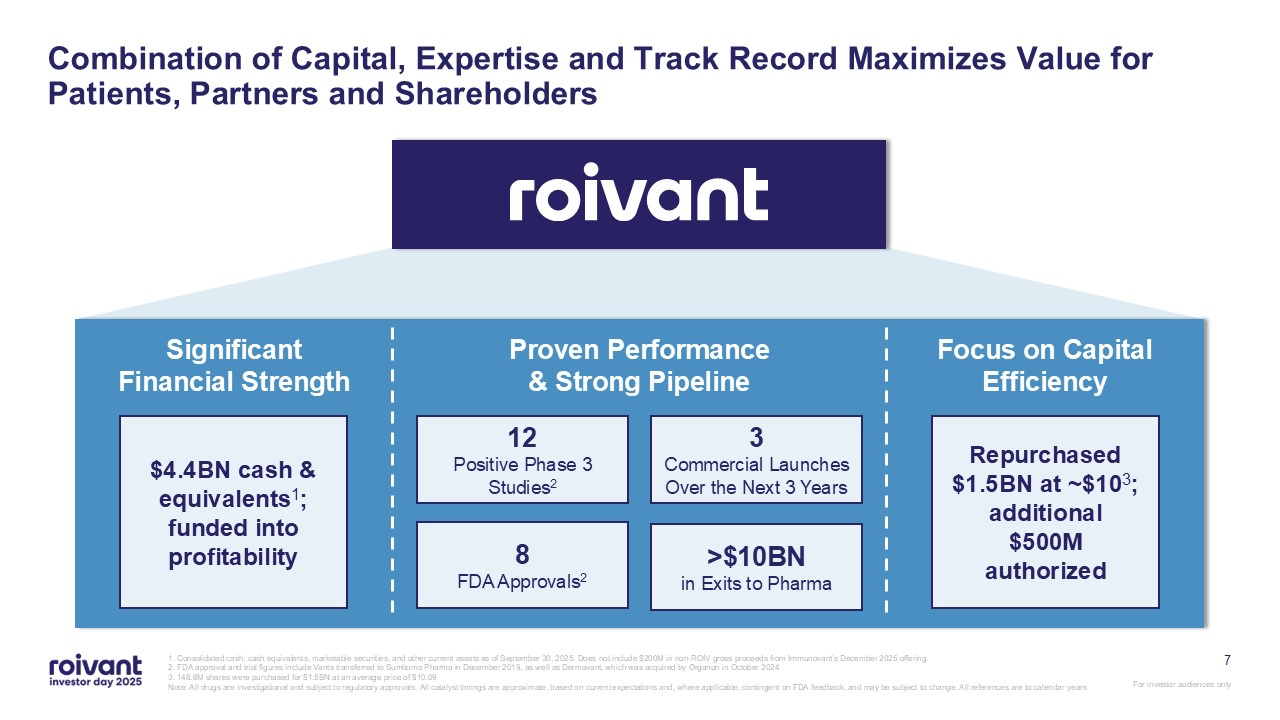
7 Combination of Capital, Expertise and Track Record Maximizes Value for
Patients, Partners and Shareholders 1. Consolidated cash, cash equivalents, marketable securities, and other current assets as of September 30, 2025. Does not include $200M in non-ROIV gross proceeds from Immunovant’s December 2025 offering.
2. FDA approval and trial figures include Vants transferred to Sumitomo Pharma in December 2019, as well as Dermavant, which was acquired by Organon in October 2024 3. 148.6M shares were purchased for $1.5BN at an average price of
$10.09 Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All
references are to calendar years Significant Financial Strength Proven Performance & Strong Pipeline Focus on Capital Efficiency 8FDA Approvals2 12Positive Phase 3 Studies2 3Commercial Launches Over the Next 3
Years >$10BNin Exits to Pharma $4.4BN cash & equivalents1; funded into profitability Repurchased $1.5BN at ~$103; additional $500M authorized
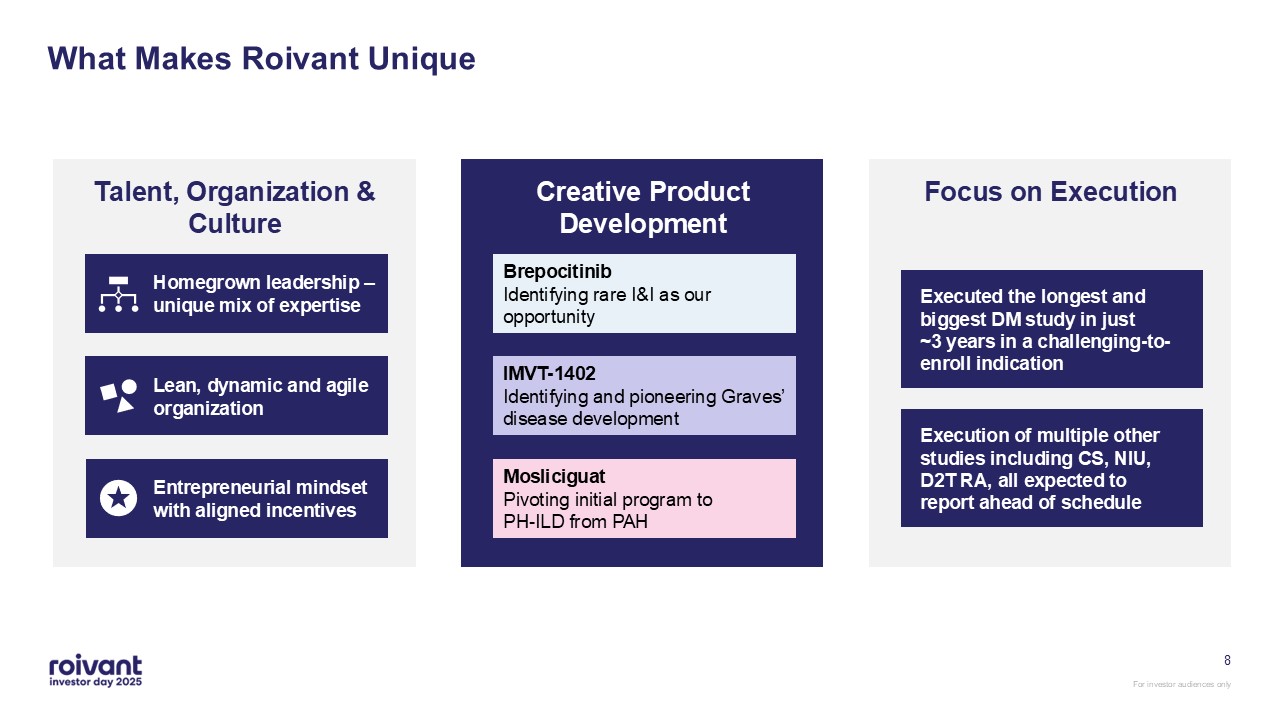
8 What Makes Roivant Unique Entrepreneurial mindset with aligned
incentives Lean, dynamic and agile organization Homegrown leadership – unique mix of expertise Talent, Organization & Culture Creative Product Development Brepocitinib Identifying rare I&I as our
opportunity IMVT-1402 Identifying and pioneering Graves’ disease development Mosliciguat Pivoting initial program to PH-ILD from PAH Focus on Execution Executed the longest and biggest DM study in just ~3 years in a
challenging-to-enroll indication Execution of multiple other studies including CS, NIU, D2T RA, all expected to report ahead of schedule

9 Strong Execution With Multiple Positive Updates to Timing Guidance Note: All
drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar
years DM NDA filing now expected early 2026 previously expected 1H 2026 NIU Ph3 trial fully enrolled with topline data now expected 2H 2026 previously expected 1H 2027 CS Ph2 trial fully enrolled with topline data now expected 1H
2026 previously expected 2H 2026 D2T RA potentially registrational trial topline data now expected 2026 previously period 1 in 2026 and topline in 2027 PH-ILD Ph2b PHocus trial enrolling well with topline data expected 2H 2026 Today's
Key Updates Brepocitinib IMVT-1402 Mosliciguat Brepocitinib Brepocitinib
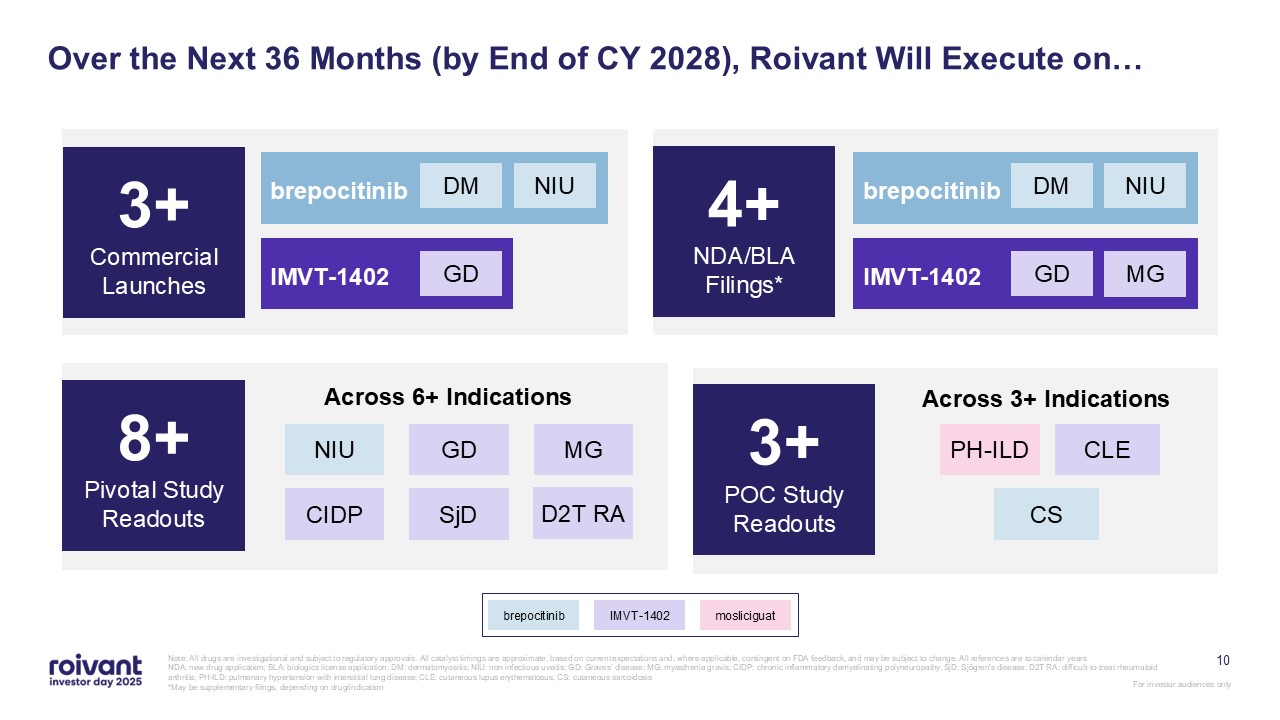
10 Over the Next 36 Months (by End of CY 2028), Roivant Will Execute on… Note:
All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to
calendar years NDA: new drug application; BLA: biologics license application; DM: dermatomyositis; NIU: non-infectious uveitis; GD: Graves’ disease; MG: myasthenia gravis; CIDP: chronic inflammatory demyelinating polyneuropathy; SjD:
Sjögren's disease; D2T RA: difficult-to-treat rheumatoid arthritis; PH-ILD: pulmonary hypertension with interstitial lung disease; CLE: cutaneous lupus erythematosus; CS: cutaneous sarcoidosis *May be supplementary filings, depending on
drug/indication 4+ NDA/BLA Filings* 8+ Pivotal Study Readouts 3+ POC Study Readouts Across 3+ Indications PH-ILD CS CLE Across 6+ Indications NIU GD MG CIDP SjD D2T RA brepocitinib IMVT-1402 GD MG NIU DM 3+ Commercial
Launches brepocitinib IMVT-1402 mosliciguat brepocitinib IMVT-1402 GD NIU DM
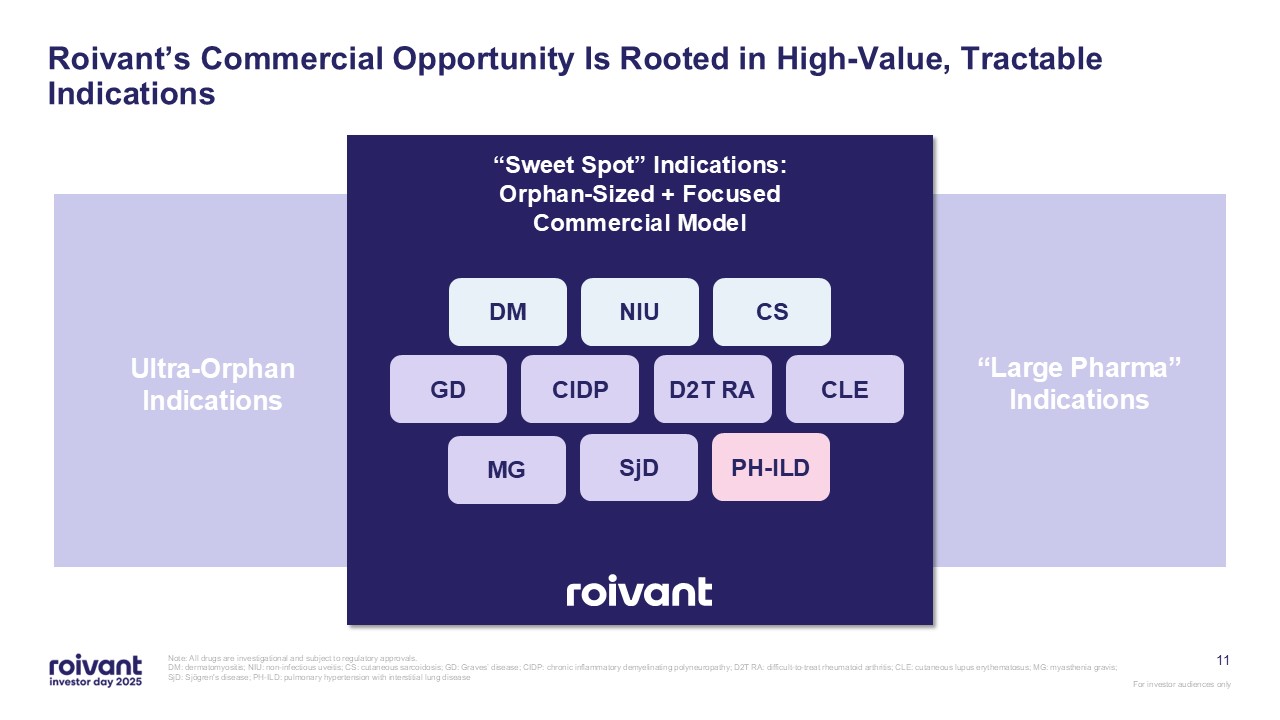
11 Roivant’s Commercial Opportunity Is Rooted in High-Value, Tractable
Indications Note: All drugs are investigational and subject to regulatory approvals. DM: dermatomyositis; NIU: non-infectious uveitis; CS: cutaneous sarcoidosis; GD: Graves’ disease; CIDP: chronic inflammatory demyelinating polyneuropathy;
D2T RA: difficult-to-treat rheumatoid arthritis; CLE: cutaneous lupus erythematosus; MG: myasthenia gravis; SjD: Sjögren's disease; PH-ILD: pulmonary hypertension with interstitial lung disease Ultra-Orphan Indications “Large Pharma”
Indications “Sweet Spot” Indications: Orphan-Sized + Focused Commercial Model DM NIU CS GD CIDP D2T RA CLE MG SjD PH-ILD
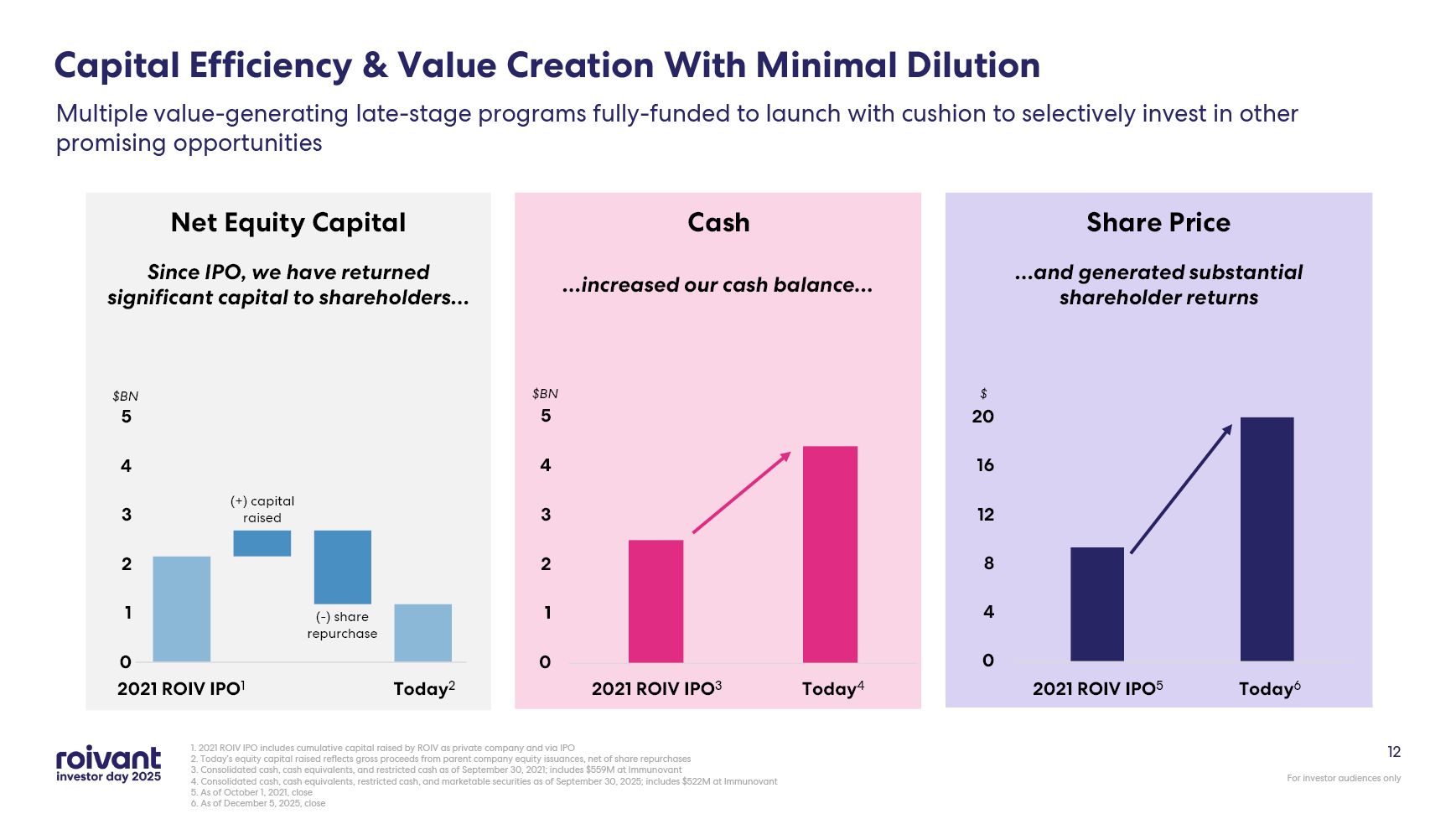
12 Capital Efficiency & Value Creation With Minimal Dilution 1. 2021 ROIV
IPO includes cumulative capital raised by ROIV as private company and via IPO 2. Today’s equity capital raised reflects gross proceeds from parent company equity issuances, net of share repurchases 3. Consolidated cash, cash equivalents,
and restricted cash as of September 30, 2021; includes $559M at Immunovant 4. Consolidated cash, cash equivalents, restricted cash, and marketable securities as of September 30, 2025; includes $522M at Immunovant 5. As of October 1, 2021,
close 6. As of December 5, 2025, close Multiple value-generating late-stage programs fully-funded to launch with cushion to selectively invest in other promising opportunities Cash Net Equity Capital Share Price $BN $ Today4 2021
ROIV IPO3 Today6 2021 ROIV IPO5 2021 ROIV IPO1 Today2 $BN Since IPO, we have returned significant capital to shareholders… …increased our cash balance… …and generated substantial shareholder returns (+) capital raised (-) share
repurchase
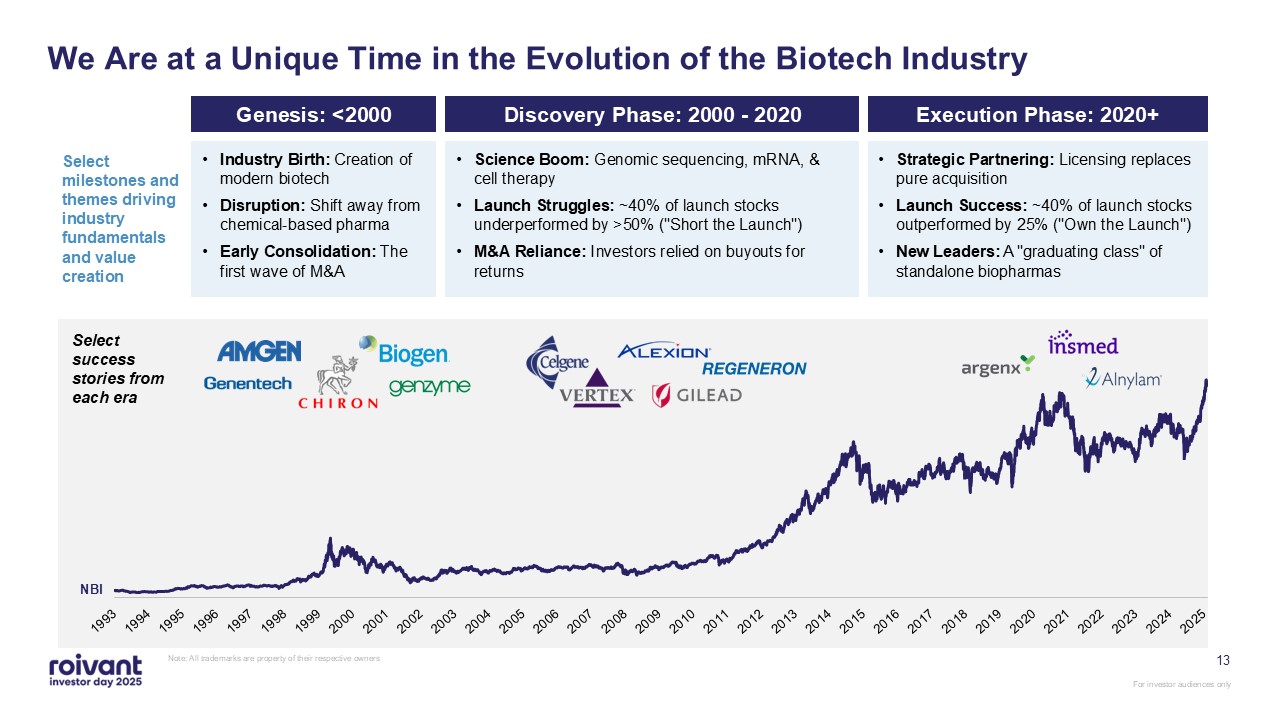
Select success stories from each era 13 We Are at a Unique Time in the
Evolution of the Biotech Industry Note: All trademarks are property of their respective owners Genesis: <2000 Strategic Partnering: Licensing replaces pure acquisition Launch Success: ~40% of launch stocks outperformed by 25% ("Own the
Launch") New Leaders: A "graduating class" of standalone biopharmas Select milestones and themes driving industry fundamentals and value creation NBI Science Boom: Genomic sequencing, mRNA, & cell therapy Launch Struggles: ~40% of
launch stocks underperformed by >50% ("Short the Launch") M&A Reliance: Investors relied on buyouts for returns Industry Birth: Creation of modern biotech Disruption: Shift away from chemical-based pharma Early Consolidation: The
first wave of M&A Discovery Phase: 2000 - 2020 Execution Phase: 2020+
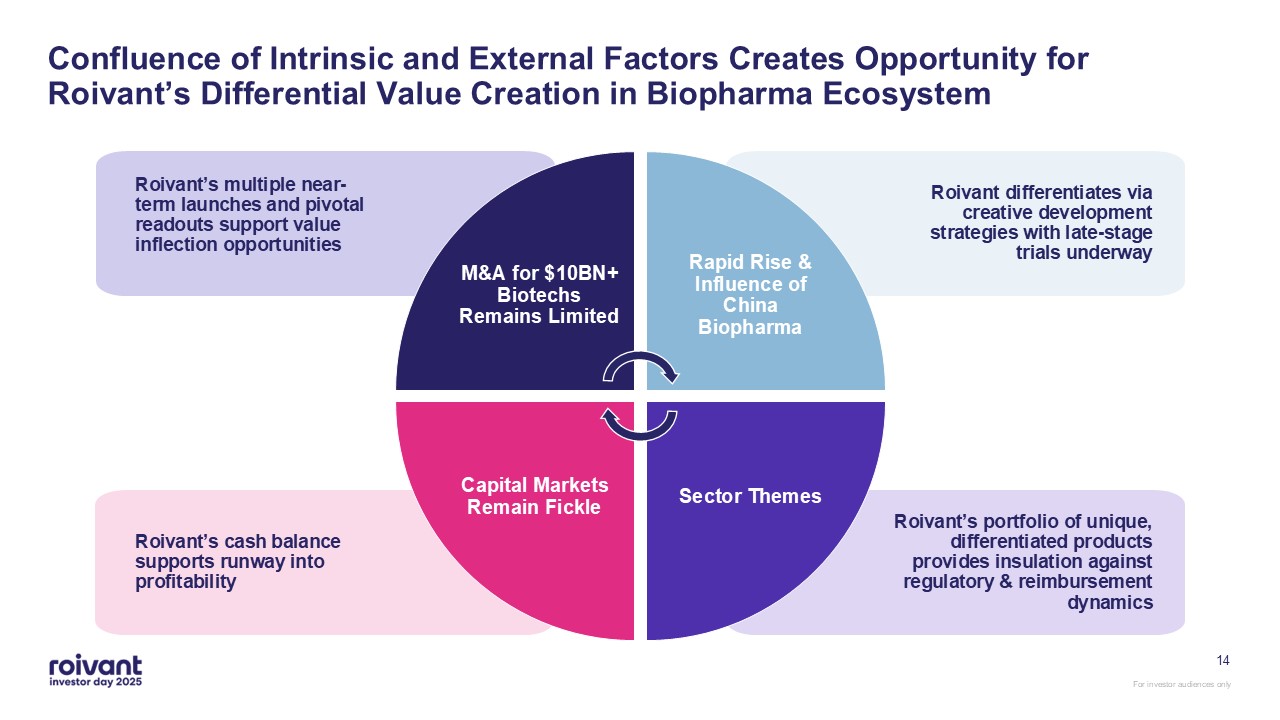
14 M&A for $10BN+ Biotechs Remains Limited Rapid Rise & Influence of
China Biopharma Sector Themes Capital Markets Remain Fickle Roivant’s portfolio of unique, differentiated products provides insulation against regulatory & reimbursement dynamics Roivant’s cash balance supports runway into
profitability Roivant’s multiple near-term launches and pivotal readouts support value inflection opportunities Roivant differentiates via creative development strategies with late-stage trials underway Confluence of Intrinsic and
External Factors Creates Opportunity for Roivant’s Differential Value Creation in Biopharma Ecosystem

15 Significant Upside and Value Creation Across Recent Launches 1. Table
represents change from post readout/pre-approval relative to today, as of December 9, 2025 2. Historical and current consensus revenue estimates sourced from Bloomberg Selected Paradigm-Shifting Pivotal Readouts Efgartigimod in gMG ADAPT
study Vutrisiran in ATTR-CM HELIOS-B study Brensocatib in NCFB ASPEN study ∆ in 2029 Consensus Rev. Estimate2 ∆ in Share Price ∆ in Market Cap +90% +204% $14BN $55BN +87% +75% $30BN $55BN + 88% +190% $11BN
$42BN ARGX ALNY INSM Rapid adoption New therapeutic options + better diagnostics grows identified prevalence Significant unmet medical need supports market access Post readout, pre-approval to today1

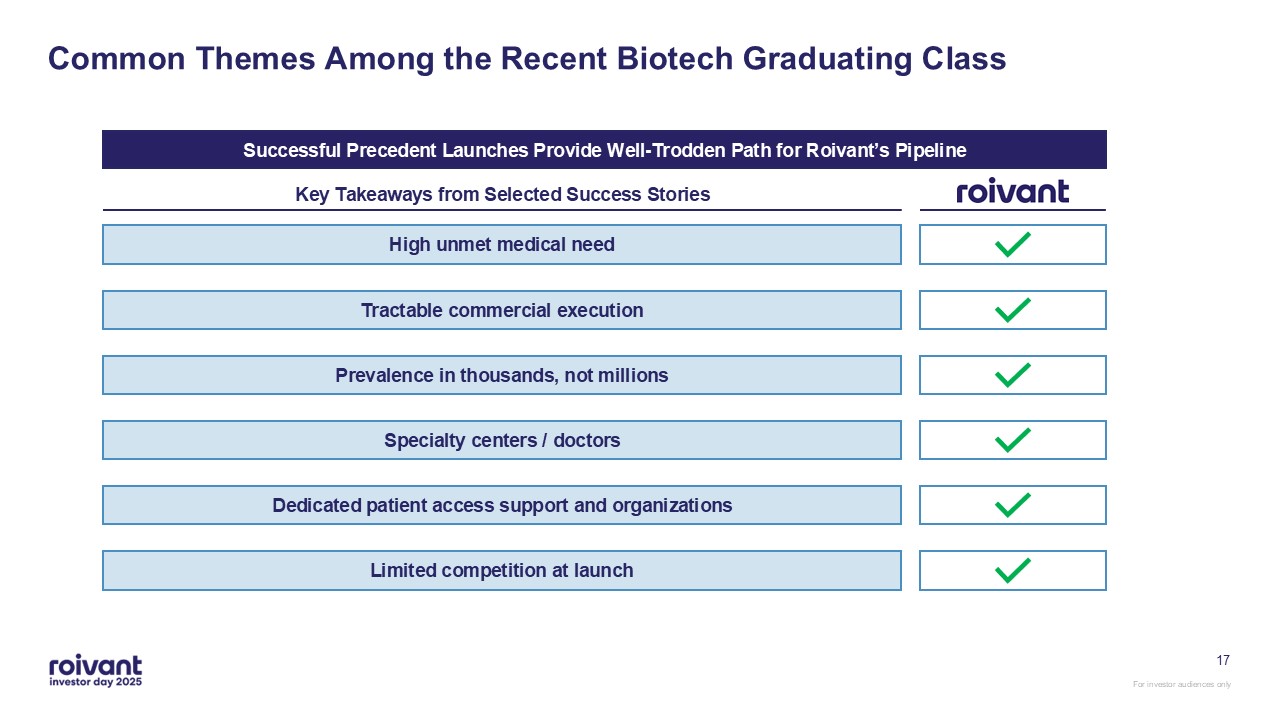
16 Common Themes Among the Recent Biotech Graduating Class Key Takeaways from
Selected Success Stories High unmet medical need Limited competition at launch Dedicated patient access support and organizations Specialty centers / doctors Prevalence in thousands, not millions Tractable commercial
execution Successful Precedent Launches Provide Well-Trodden Path for Roivant’s Pipeline
17 Common Themes Among the Recent Biotech Graduating Class Key Takeaways from
Selected Success Stories High unmet medical need Limited competition at launch Dedicated patient access support and organizations Specialty centers / doctors Prevalence in thousands, not millions Tractable commercial
execution Successful Precedent Launches Provide Well-Trodden Path for Roivant’s Pipeline
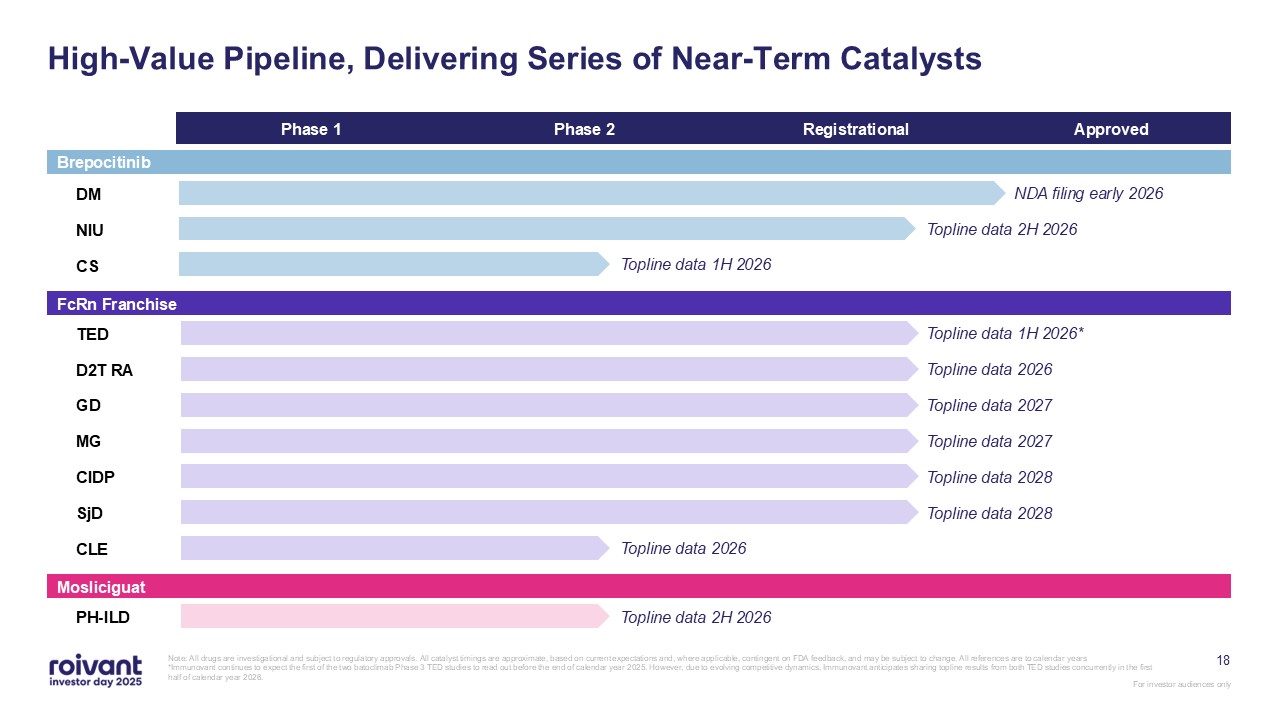
18 Phase 1 Phase 2 Registrational Approved DM NIU CS TED D2T
RA GD MG CIDP SjD CLE PH-ILD High-Value Pipeline, Delivering Series of Near-Term Catalysts Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations
and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years *Immunovant continues to expect the first of the two batoclimab Phase 3 TED studies to read out before the end of calendar
year 2025. However, due to evolving competitive dynamics, Immunovant anticipates sharing topline results from both TED studies concurrently in the first half of calendar year 2026. NDA filing early 2026 Topline data 2H 2026 Topline data 1H
2026 Topline data 2026 Topline data 2027 Topline data 2027 Topline data 2028 Topline data 2028 Topline data 1H 2026* Topline data 2026 Topline data 2H 2026 FcRn Franchise Mosliciguat Brepocitinib

19 Brepocitinib Ben Zimmer CEO, Priovant Matt Gline CEO, Roivant

20 Brepocitinib program is focused on indications with biology suited for dual
JAK1/TYK2 inhibition and significant unmet need NDA filing for brepocitinib in DM expected in early 2026; preparations underway for potential commercial launch in DM in early 2027 No approved therapies and risk of permanent cutaneous
damage highlight unmet need in CS; topline data from Phase 2 BEACON study expected to read out 1H 2026 ahead of prior guidance (2H 2026) NIU treatment paradigm enables potential for new therapeutic uptake across market segments; topline data
from Phase 3 CLARITY study expected to read out 2H 2026 ahead of prior guidance (1H 2027) DM standard of care leaves patients poorly controlled, dissatisfied, and exposed to high steroid burden, underscoring the need for new treatments Key
Takeaways: Brepocitinib NIU: non-infectious uveitis; CS: cutaneous sarcoidosis; DM: dermatomyositis Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations
and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years. For investor audiences only
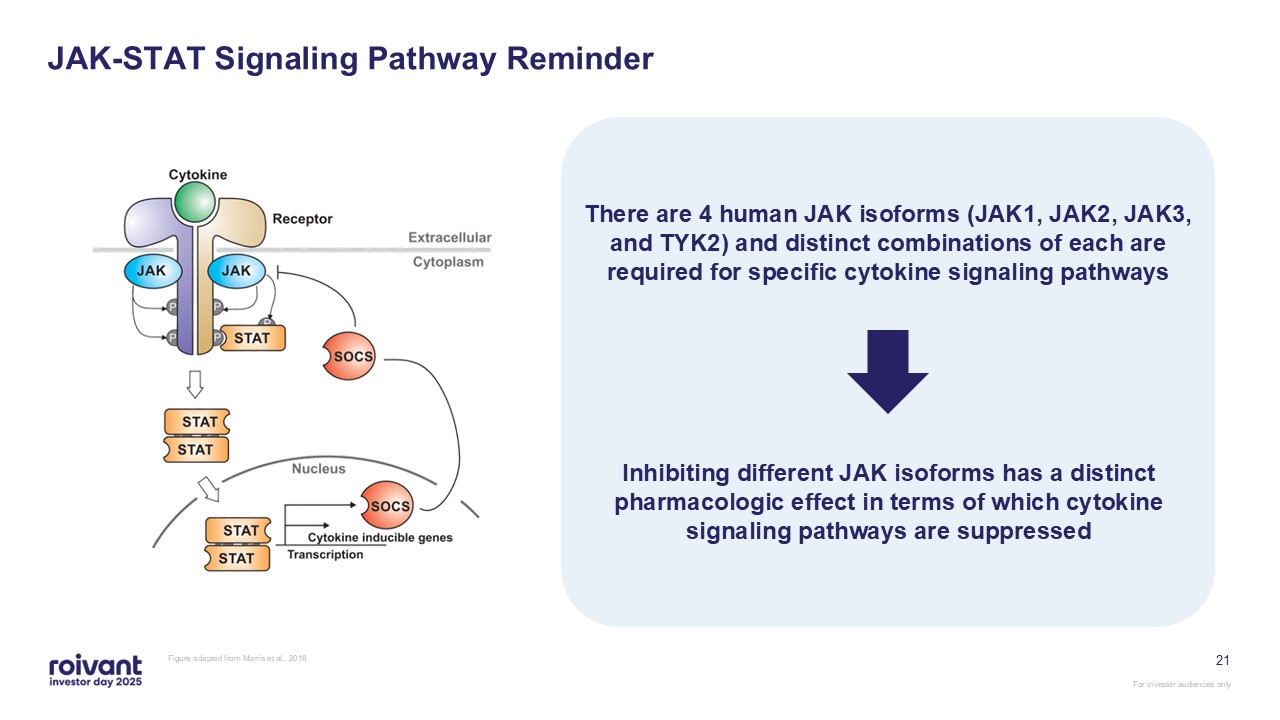
JAK-STAT Signaling Pathway Reminder Figure adapted from Morris et al.,
2018 21 There are 4 human JAK isoforms (JAK1, JAK2, JAK3, and TYK2) and distinct combinations of each are required for specific cytokine signaling pathways Inhibiting different JAK isoforms has a distinct pharmacologic effect in terms of
which cytokine signaling pathways are suppressed
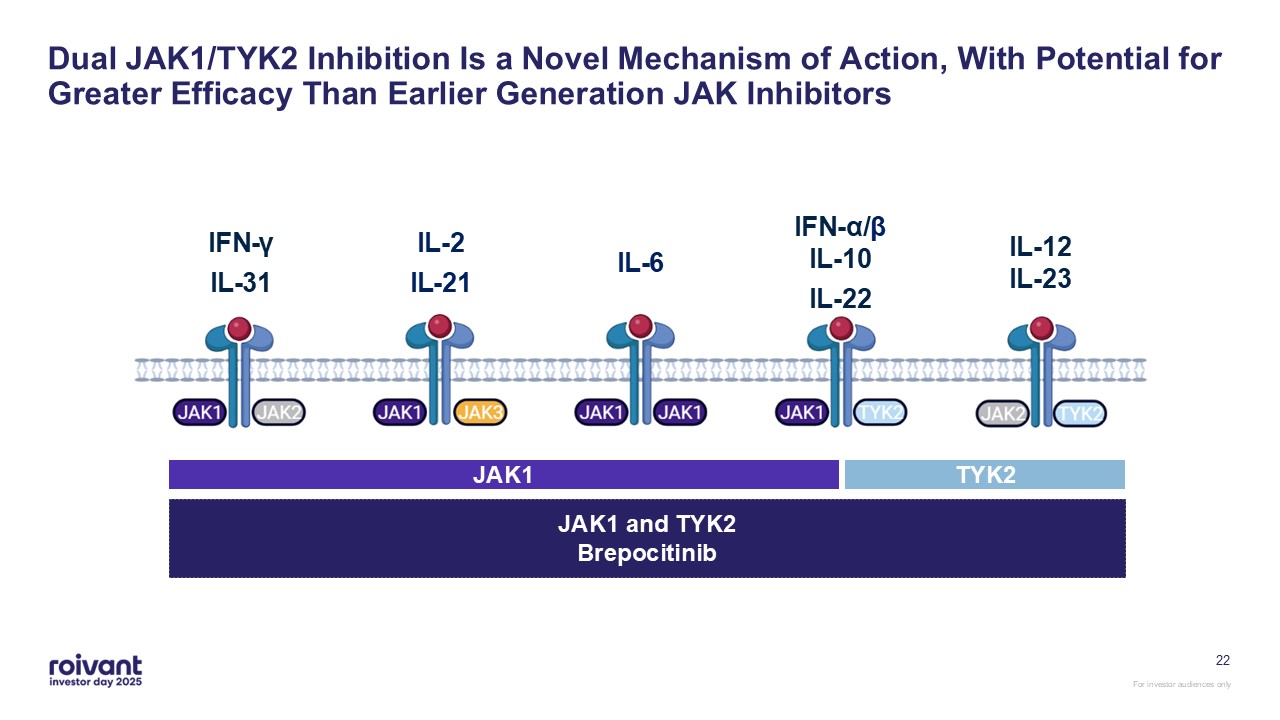
Dual JAK1/TYK2 Inhibition Is a Novel Mechanism of Action, With Potential for
Greater Efficacy Than Earlier Generation JAK Inhibitors IFN-γ IL-31 IL-2 IL-21 IL-6 IFN-α/βIL-10 IL-22 IL-12IL-23 JAK1 and TYK2Brepocitinib JAK1 TYK2 22
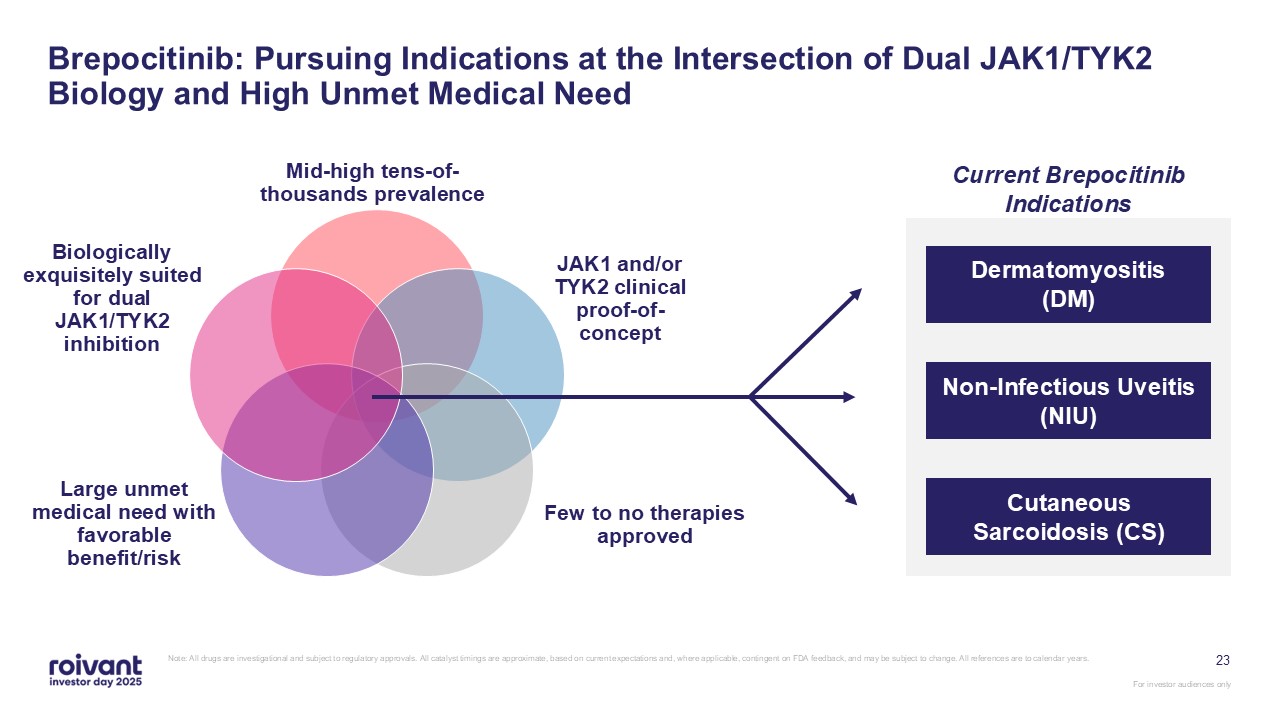
23 Brepocitinib: Pursuing Indications at the Intersection of Dual JAK1/TYK2
Biology and High Unmet Medical Need Non-Infectious Uveitis, Dermatomyositis, Cutaneous Sarcoidosis Mid-high tens-of-thousands prevalence JAK1 and/or TYK2 clinical proof-of-concept Few to no therapies approved Large unmet medical need
with favorable benefit/risk Biologically exquisitely suited for dual JAK1/TYK2 inhibition Non-Infectious Uveitis (NIU) Dermatomyositis (DM) Cutaneous Sarcoidosis (CS) Current Brepocitinib Indications Note: All drugs are
investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years.
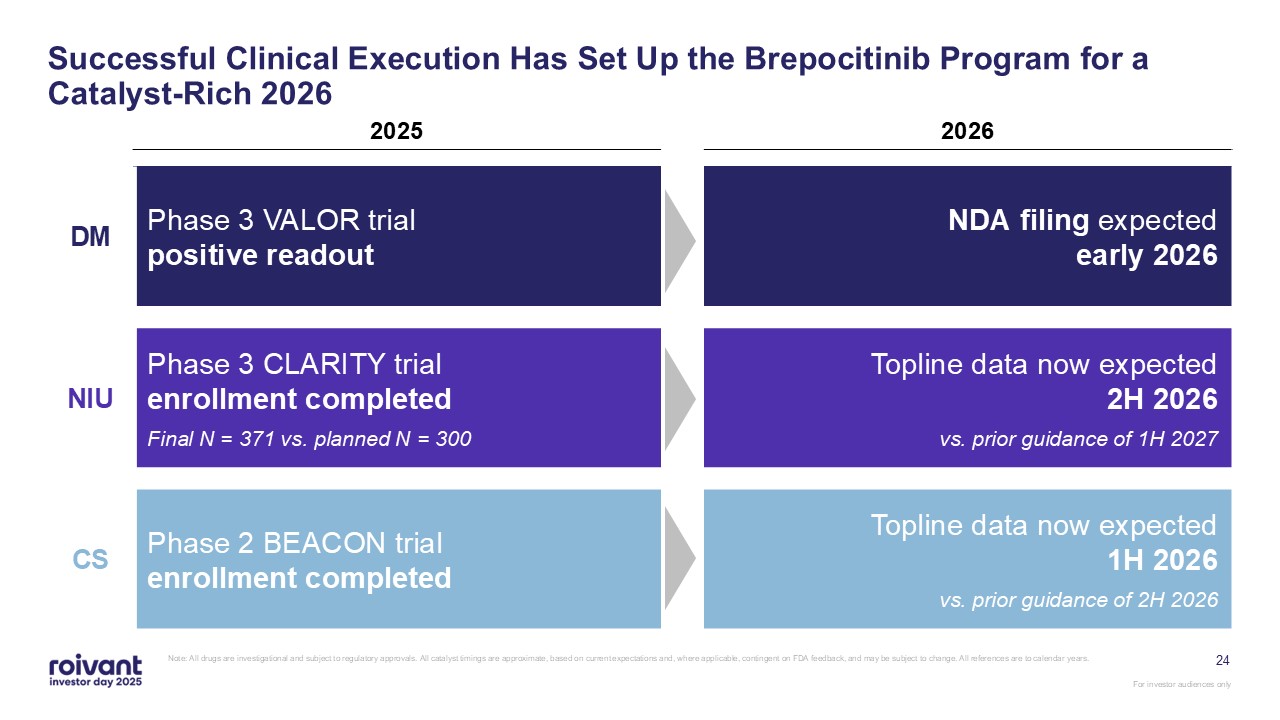
24 Successful Clinical Execution Has Set Up the Brepocitinib Program for a
Catalyst-Rich 2026 Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change.
All references are to calendar years. 2025 2026 DM Phase 3 VALOR trial positive readout NDA filing expected early 2026 NIU Phase 3 CLARITY trial enrollment completed Final N = 371 vs. planned N = 300 Topline data now expected 2H
2026 vs. prior guidance of 1H 2027 CS Phase 2 BEACON trial enrollment completed Topline data now expected 1H 2026 vs. prior guidance of 2H 2026

Non-Infectious Uveitis
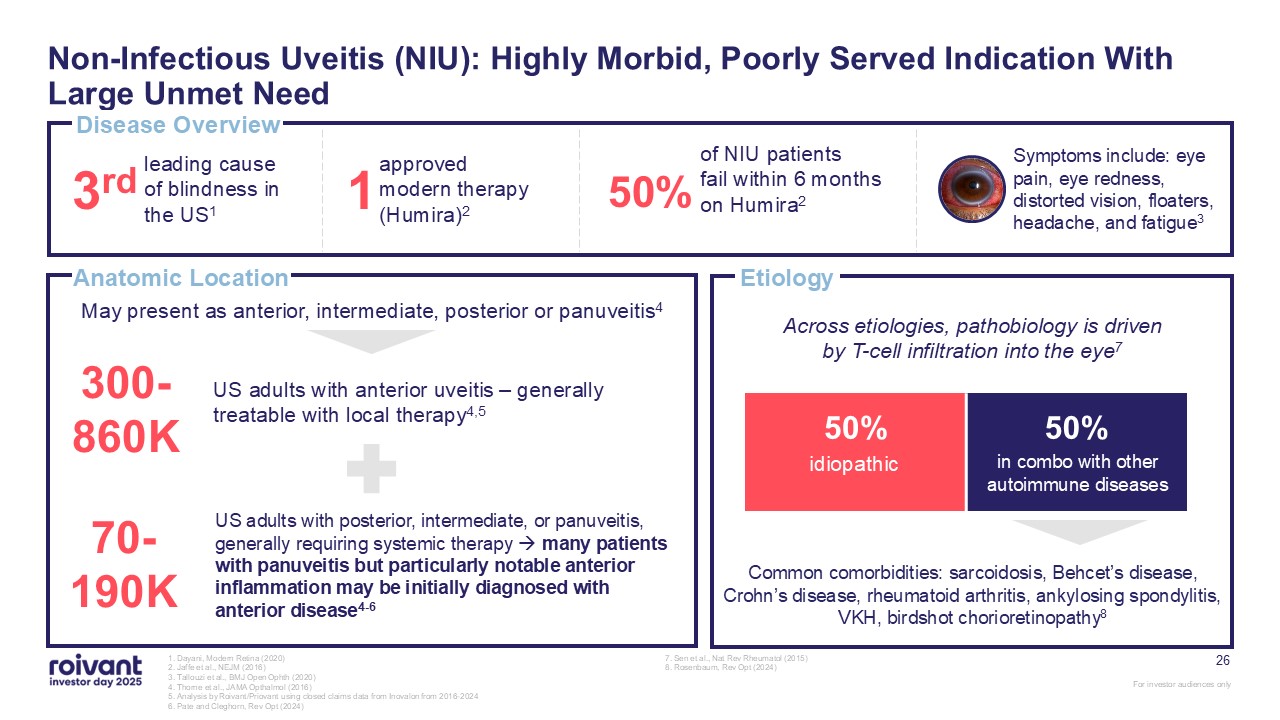
26 Non-Infectious Uveitis (NIU): Highly Morbid, Poorly Served Indication With
Large Unmet Need 1. Dayani, Modern Retina (2020) 2. Jaffe et al., NEJM (2016) 3. Tallouzi et al., BMJ Open Ophth (2020) 4. Thorne et al., JAMA Opthalmol (2016) 5. Analysis by Roivant/Priovant using closed claims data from Inovalon from
2016-2024 6. Pate and Cleghorn, Rev Opt (2024) 7. Sen et al., Nat Rev Rheumatol (2015) 8. Rosenbaum, Rev Opt (2024) Disease Overview Anatomic Location 3rd leading cause of blindness in the US1 1 approved modern therapy
(Humira)2 50% of NIU patients fail within 6 months on Humira2 300-860K US adults with anterior uveitis – generally treatable with local therapy4,5 70-190K US adults with posterior, intermediate, or panuveitis, generally requiring
systemic therapy many patients with panuveitis but particularly notable anterior inflammation may be initially diagnosed with anterior disease4-6 May present as anterior, intermediate, posterior or panuveitis4 Etiology 50% 50% Common
comorbidities: sarcoidosis, Behcet’s disease, Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, VKH, birdshot chorioretinopathy8 idiopathic in combo with other autoimmune diseases Across etiologies, pathobiology is driven by
T-cell infiltration into the eye7 Symptoms include: eye pain, eye redness, distorted vision, floaters, headache, and fatigue3
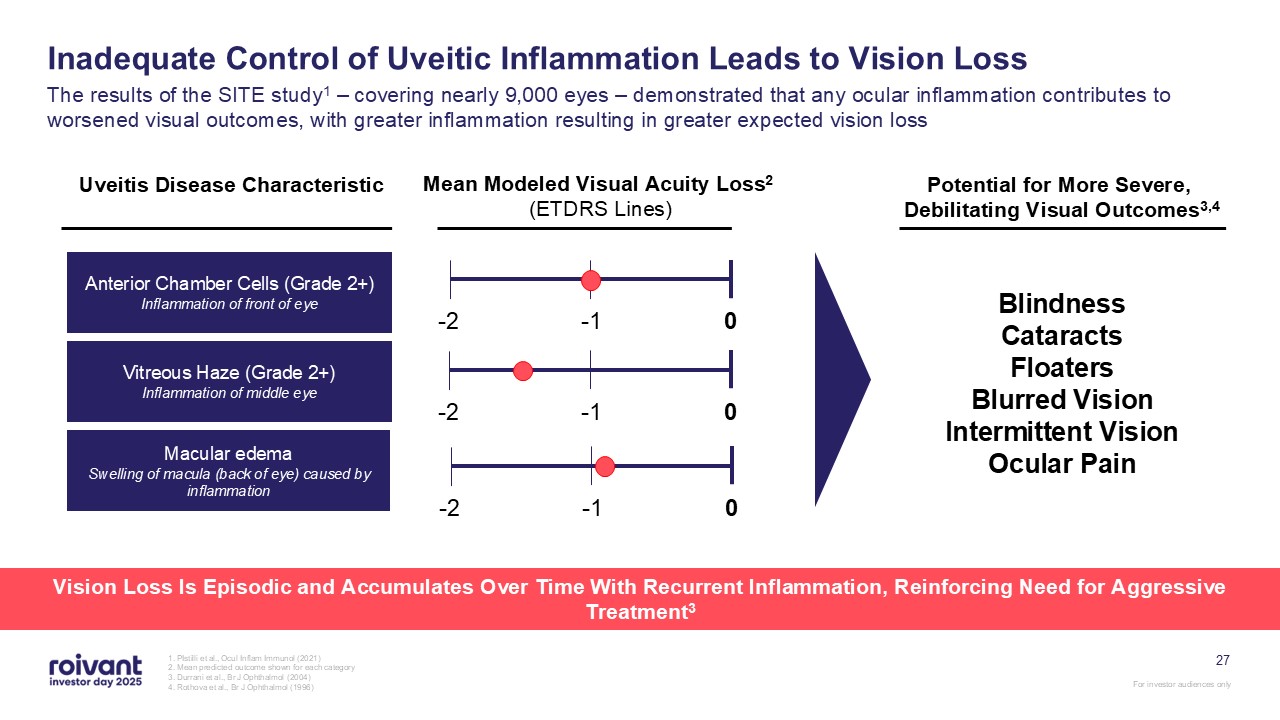
27 Inadequate Control of Uveitic Inflammation Leads to Vision Loss The results
of the SITE study1 – covering nearly 9,000 eyes – demonstrated that any ocular inflammation contributes to worsened visual outcomes, with greater inflammation resulting in greater expected vision loss 1. PIstilli et al., Ocul Inflam Immunol
(2021) 2. Mean predicted outcome shown for each category 3. Durrani et al., Br J Ophthalmol (2004) 4. Rothova et al., Br J Ophthalmol (1996) Anterior Chamber Cells (Grade 2+) Inflammation of front of eye Vitreous Haze (Grade
2+) Inflammation of middle eye Macular edema Swelling of macula (back of eye) caused by inflammation 0 -2 -1 Uveitis Disease Characteristic Mean Modeled Visual Acuity Loss2 (ETDRS Lines) Potential for More Severe, Debilitating
Visual Outcomes3,4 Blindness Cataracts Floaters Blurred Vision Intermittent Vision Ocular Pain 0 -2 -1 0 -2 -1 Vision Loss Is Episodic and Accumulates Over Time With Recurrent Inflammation, Reinforcing Need for Aggressive
Treatment3
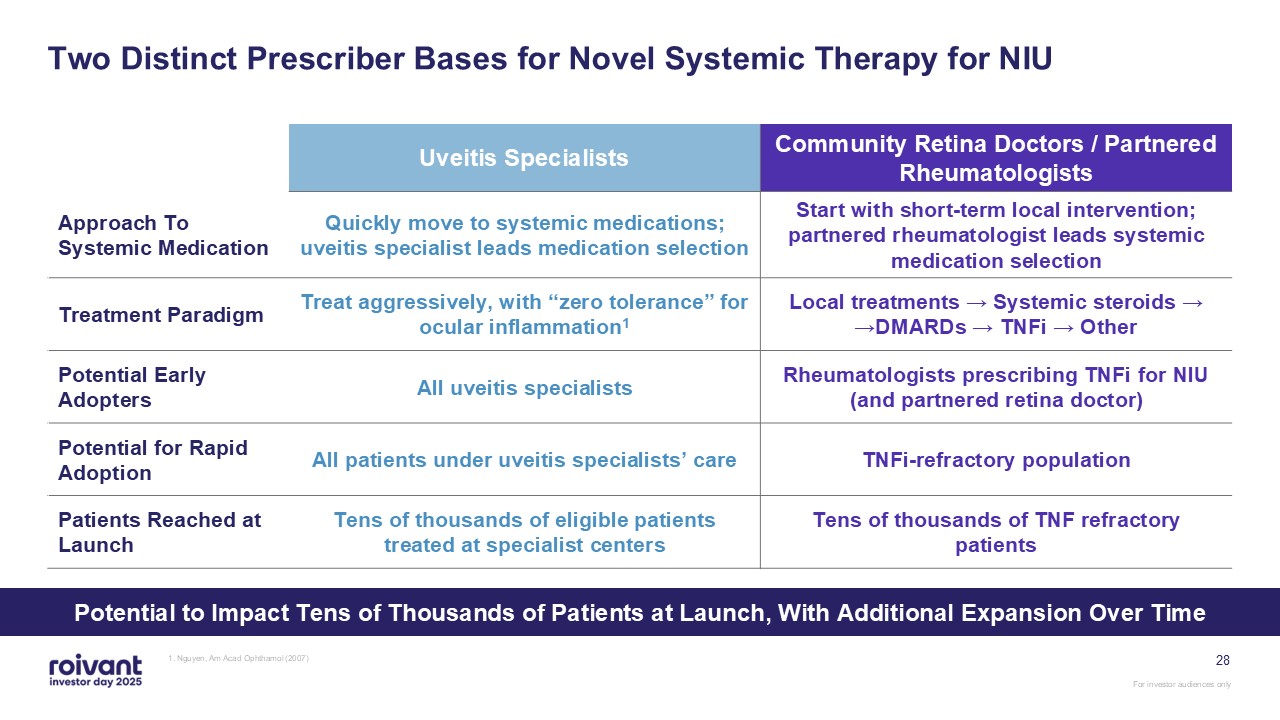
28 Two Distinct Prescriber Bases for Novel Systemic Therapy for NIU Uveitis
Specialists Community Retina Doctors / Partnered Rheumatologists Approach To Systemic Medication Quickly move to systemic medications; uveitis specialist leads medication selection Start with short-term local intervention; partnered
rheumatologist leads systemic medication selection Treatment Paradigm Treat aggressively, with “zero tolerance” for ocular inflammation1 Local treatments → Systemic steroids → →DMARDs → TNFi → Other Potential Early Adopters All uveitis
specialists Rheumatologists prescribing TNFi for NIU (and partnered retina doctor) Potential for Rapid Adoption All patients under uveitis specialists’ care TNFi-refractory population Patients Reached at Launch Tens of thousands of
eligible patients treated at specialist centers Tens of thousands of TNF refractory patients 1. Nguyen, Am Acad Ophthamol (2007) Potential to Impact Tens of Thousands of Patients at Launch, With Additional Expansion Over Time
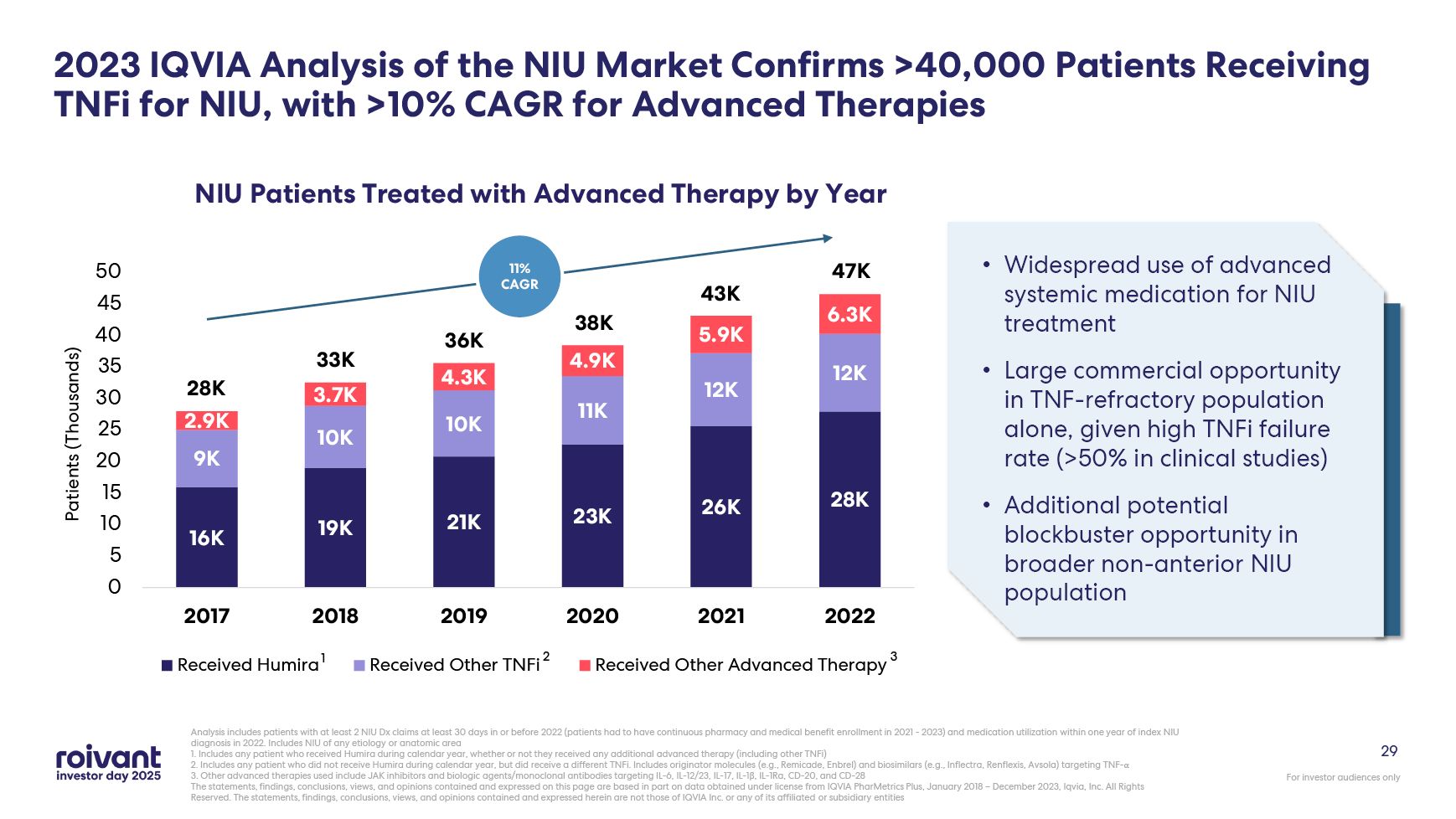
29 2023 IQVIA Analysis of the NIU Market Confirms >40,000 Patients Receiving
TNFi for NIU, with >10% CAGR for Advanced Therapies Analysis includes patients with at least 2 NIU Dx claims at least 30 days in or before 2022 (patients had to have continuous pharmacy and medical benefit enrollment in 2021 - 2023) and
medication utilization within one year of index NIU diagnosis in 2022. Includes NIU of any etiology or anatomic area 1. Includes any patient who received Humira during calendar year, whether or not they received any additional advanced
therapy (including other TNFi) 2. Includes any patient who did not receive Humira during calendar year, but did receive a different TNFi. Includes originator molecules (e.g., Remicade, Enbrel) and biosimilars (e.g., Inflectra, Renflexis,
Avsola) targeting TNF-α 3. Other advanced therapies used include JAK inhibitors and biologic agents/monoclonal antibodies targeting IL-6, IL-12/23, IL-17, IL-1β, IL-1Ra, CD-20, and CD-28 The statements, findings, conclusions, views, and
opinions contained and expressed on this page are based in part on data obtained under license from IQVIA PharMetrics Plus, January 2018 – December 2023, Iqvia, Inc. All Rights Reserved. The statements, findings, conclusions, views, and
opinions contained and expressed herein are not those of IQVIA Inc. or any of its affiliated or subsidiary entities Widespread use of advanced systemic medication for NIU treatment Large commercial opportunity in TNF-refractory population
alone, given high TNFi failure rate (>50% in clinical studies) Additional potential blockbuster opportunity in broader non-anterior NIU population 28K 33K 36K 38K 43K 47K 11% CAGR 1 2 3
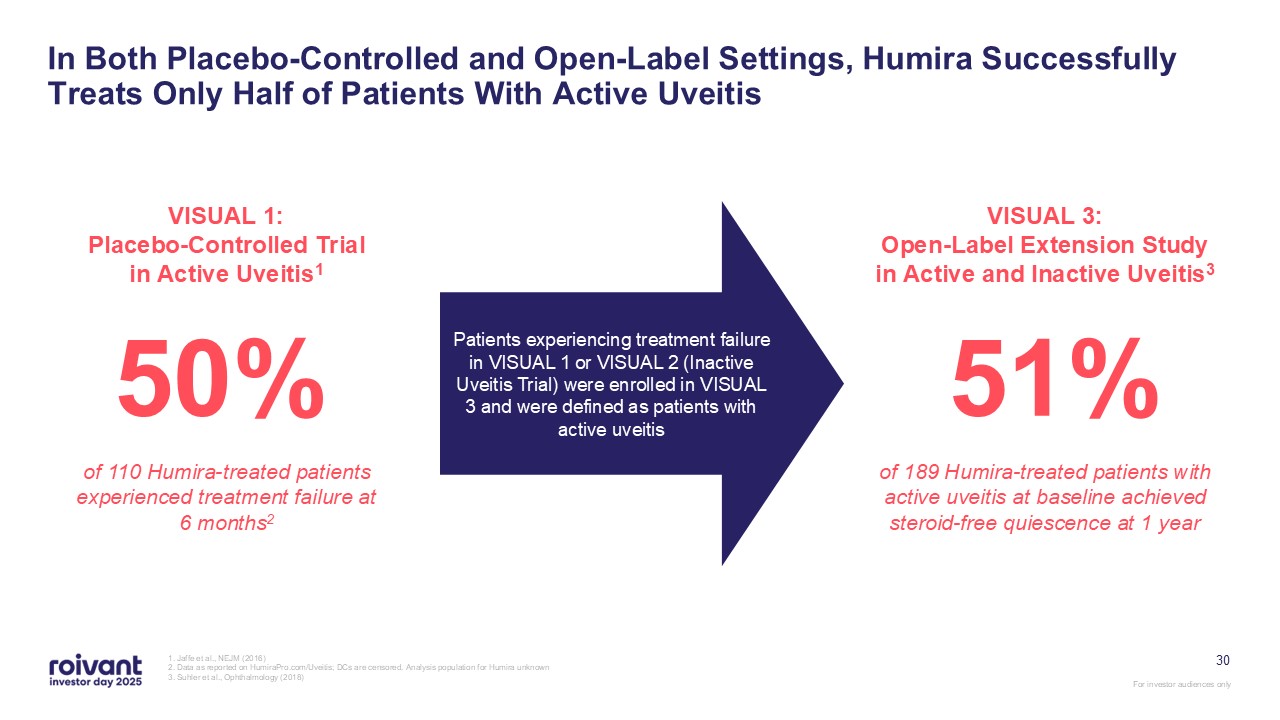
30 In Both Placebo-Controlled and Open-Label Settings, Humira Successfully
Treats Only Half of Patients With Active Uveitis 1. Jaffe et al., NEJM (2016) 2. Data as reported on HumiraPro.com/Uveitis; DCs are censored. Analysis population for Humira unknown 3. Suhler et al., Ophthalmology (2018) VISUAL 1:
Placebo-Controlled Trial in Active Uveitis1 50% of 110 Humira-treated patients experienced treatment failure at 6 months2 VISUAL 3: Open-Label Extension Study in Active and Inactive Uveitis3 51% of 189 Humira-treated patients with
active uveitis at baseline achieved steroid-free quiescence at 1 year Patients experiencing treatment failure in VISUAL 1 or VISUAL 2 (Inactive Uveitis Trial) were enrolled in VISUAL 3 and were defined as patients with active uveitis
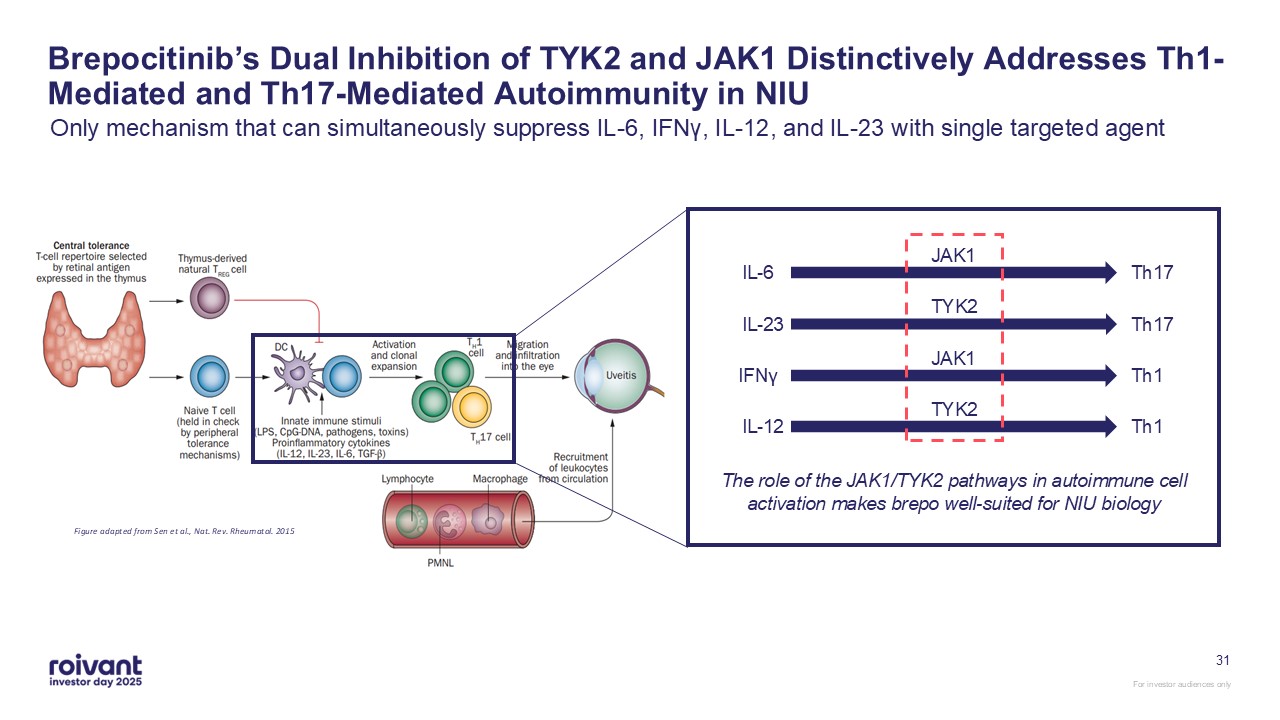
31 Brepocitinib’s Dual Inhibition of TYK2 and JAK1 Distinctively Addresses
Th1-Mediated and Th17-Mediated Autoimmunity in NIU Only mechanism that can simultaneously suppress IL-6, IFNγ, IL-12, and IL-23 with single targeted agent Figure adapted from Sen et al., Nat. Rev. Rheumatol.
2015 JAK1 IL-6 Th17 TYK2 IL-23 Th17 JAK1 IFNγ Th1 TYK2 IL-12 Th1 The role of the JAK1/TYK2 pathways in autoimmune cell activation makes brepo well-suited for NIU biology

32 NEPTUNE: Phase 2 Study of Brepocitinib in NIU Note: Additional inclusion
and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide QD: daily; OCS: oral corticosteroids; ACC: anterior chamber cell; VH: vitreous haze; BCVA: Best Corrected Visual Acuity
References are to calendar years Positive readout in 2024 N = 26 Adults with active non-infectious intermediate-, posterior-, or panuveitis Brepocitinib 15 mg QD Brepocitinib 45 mg QD 52 Week 0 Primary Efficacy Endpoint:
Treatment Failure at Week 24 Treatment failure determined by hitting certain thresholds for increase in one or more of ACC grade VH grade Inflammatory lesions BCVA 2:1 Randomization Steroid burst and taper: 60 mg/day OCS burst for 14
days; forced taper to 0 mg/day over 6 weeks
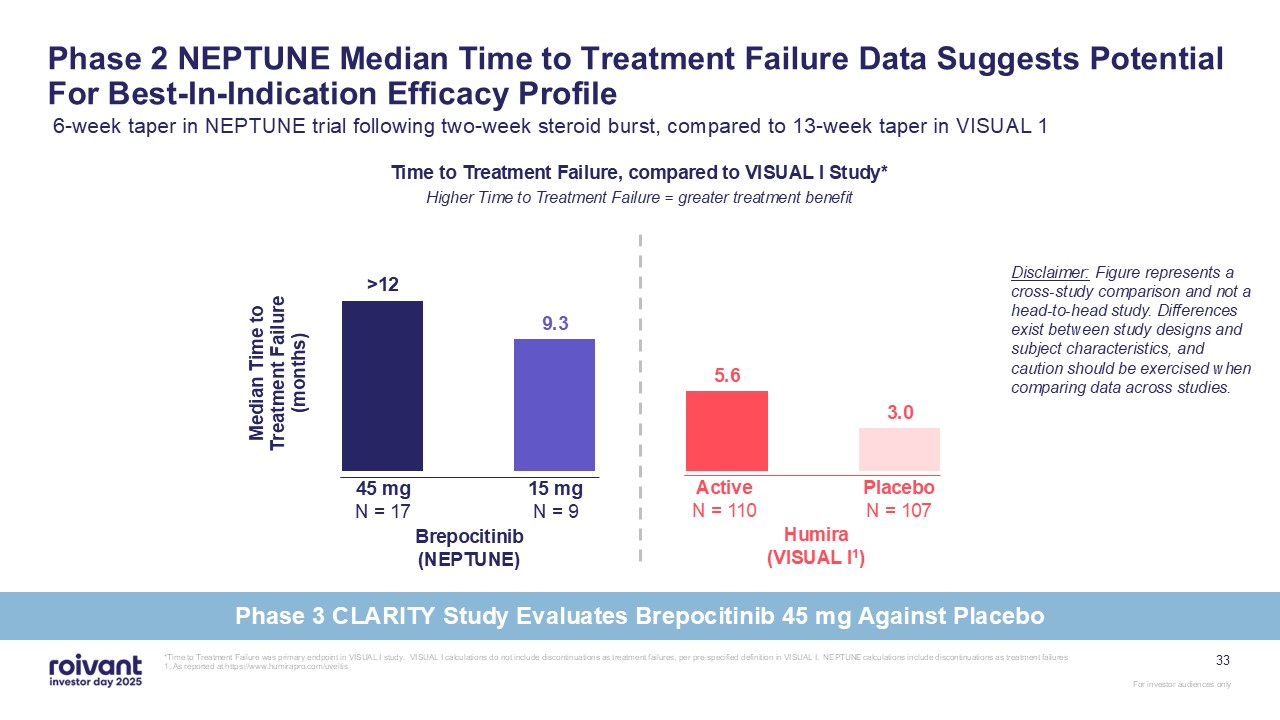
Phase 2 NEPTUNE Median Time to Treatment Failure Data Suggests Potential For
Best-In-Indication Efficacy Profile 6-week taper in NEPTUNE trial following two-week steroid burst, compared to 13-week taper in VISUAL 1 45 mg N = 17 Brepocitinib (NEPTUNE) 15 mg N = 9 Active N = 110 Humira (VISUAL
I1) Placebo N = 107 Disclaimer: Figure represents a cross-study comparison and not a head-to-head study. Differences exist between study designs and subject characteristics, and caution should be exercised when comparing data across
studies. Time to Treatment Failure, compared to VISUAL I Study* Higher Time to Treatment Failure = greater treatment benefit *Time to Treatment Failure was primary endpoint in VISUAL I study. VISUAL I calculations do not include
discontinuations as treatment failures, per pre-specified definition in VISUAL I. NEPTUNE calculations include discontinuations as treatment failures 1. As reported at https://www.humirapro.com/uveitis 33 Phase 3 CLARITY Study Evaluates
Brepocitinib 45 mg Against Placebo
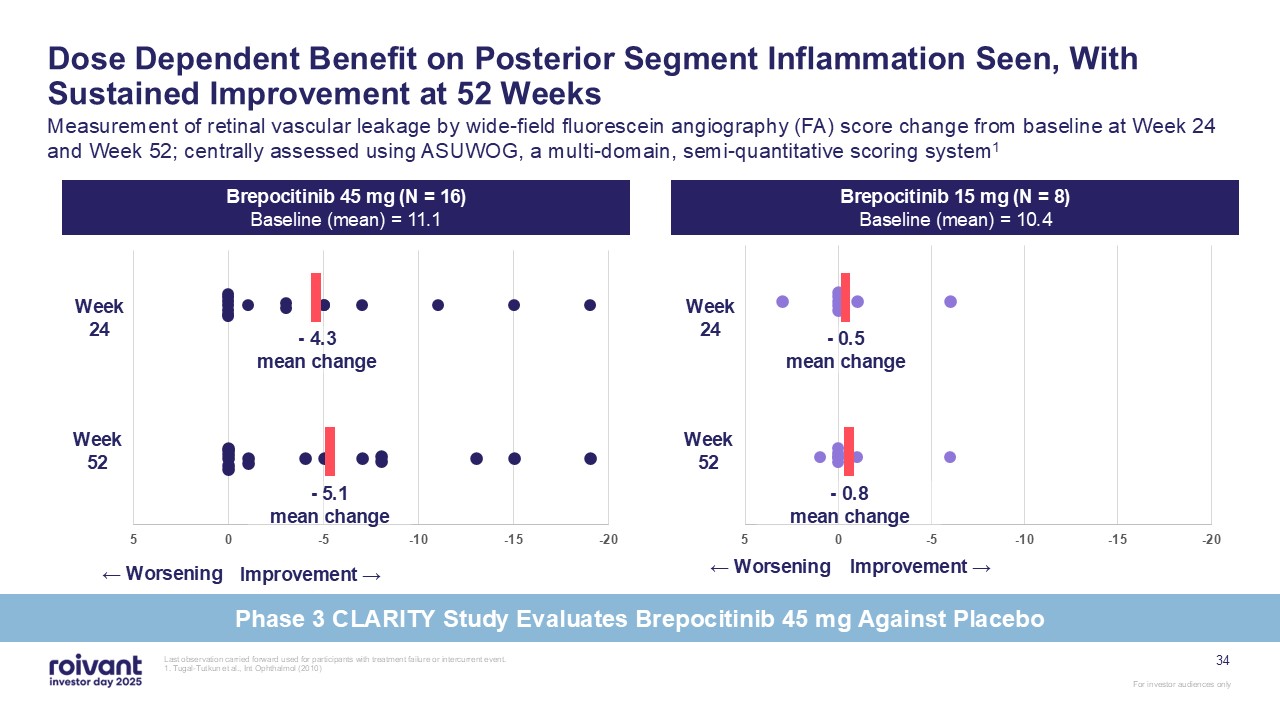
Last observation carried forward used for participants with treatment failure or
intercurrent event. 1. Tugal-Tutkun et al., Int Ophthalmol (2010) Dose Dependent Benefit on Posterior Segment Inflammation Seen, With Sustained Improvement at 52 Weeks Measurement of retinal vascular leakage by wide-field fluorescein
angiography (FA) score change from baseline at Week 24 and Week 52; centrally assessed using ASUWOG, a multi-domain, semi-quantitative scoring system1 ← Worsening Improvement → - 0.5mean change - 4.3mean change - 0.8mean change -
5.1mean change ← Worsening Improvement → Brepocitinib 45 mg (N = 16) Baseline (mean) = 11.1 Brepocitinib 15 mg (N = 8) Baseline (mean) = 10.4 Week 24 Week 52 Week 24 Week 52 34 Phase 3 CLARITY Study Evaluates Brepocitinib 45 mg
Against Placebo
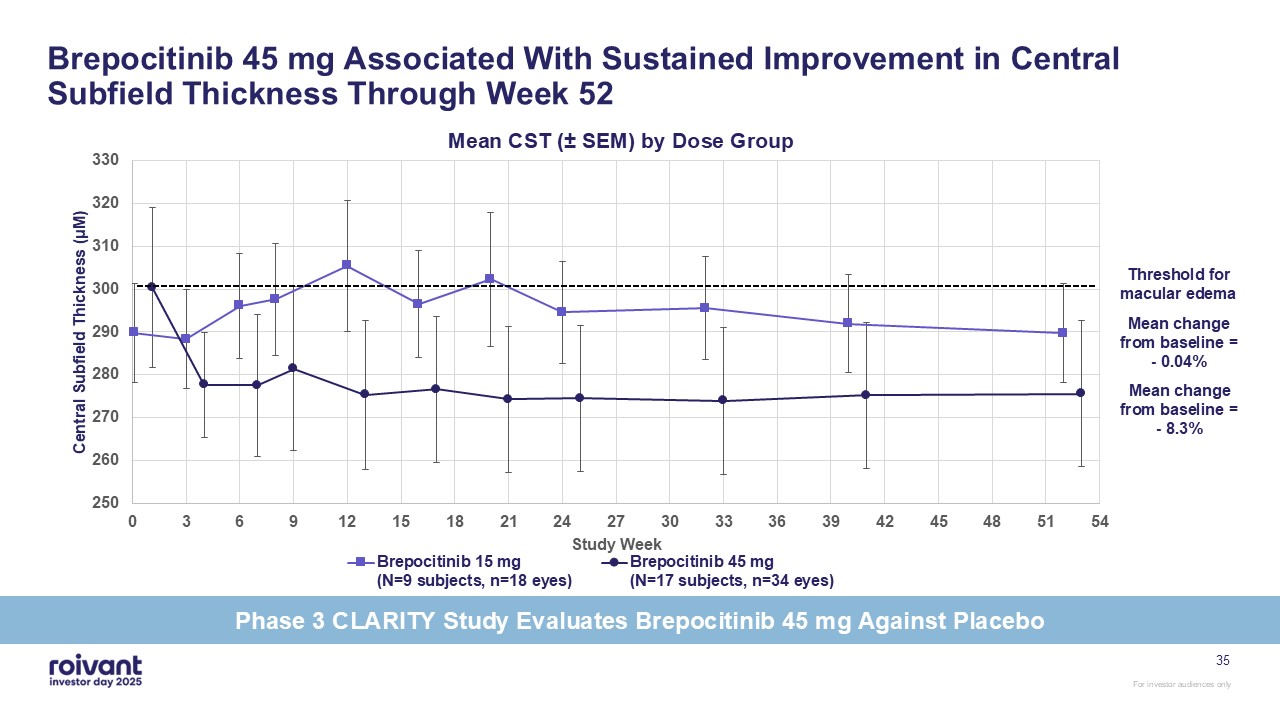
Brepocitinib 45 mg Associated With Sustained Improvement in Central Subfield
Thickness Through Week 52 35 Threshold for macular edema Mean change from baseline = - 0.04% Mean change from baseline = - 8.3% Phase 3 CLARITY Study Evaluates Brepocitinib 45 mg Against Placebo

36 CLARITY: Phase 3 Study of Brepocitinib in NIU Two identical sub-studies,
CLARITY-1 and CLARITY-2, under a single protocol; study is now fully enrolled and topline results are expected 2H 2026 Note: Additional inclusion and exclusion criteria not listed on slide. Note: Additional primary and secondary endpoint
details not listed on slide QD: daily; OCS: oral corticosteroids; CST: central subfield thickness; WFFA: wide-field fluorescein angiography References are to calendar years Global N=371 CLARITY-1 N=180 CLARITY-2 N=191 Adults with active
non-infectious intermediate-, posterior-, or panuveitis Placebo Brepocitinib 45 mg QD 48 Week 0 Primary Endpoint: Time to Treatment Failure Secondary Endpoints: Include measurements related to Macular edema/CST Retinal vascular
leakage as measured through WFFA Functional vision 1:1 Randomization Steroid burst and taper: 60 mg/day OCS burst for 14 days; forced taper to 0 mg/day over 6 weeks

Cutaneous Sarcoidosis
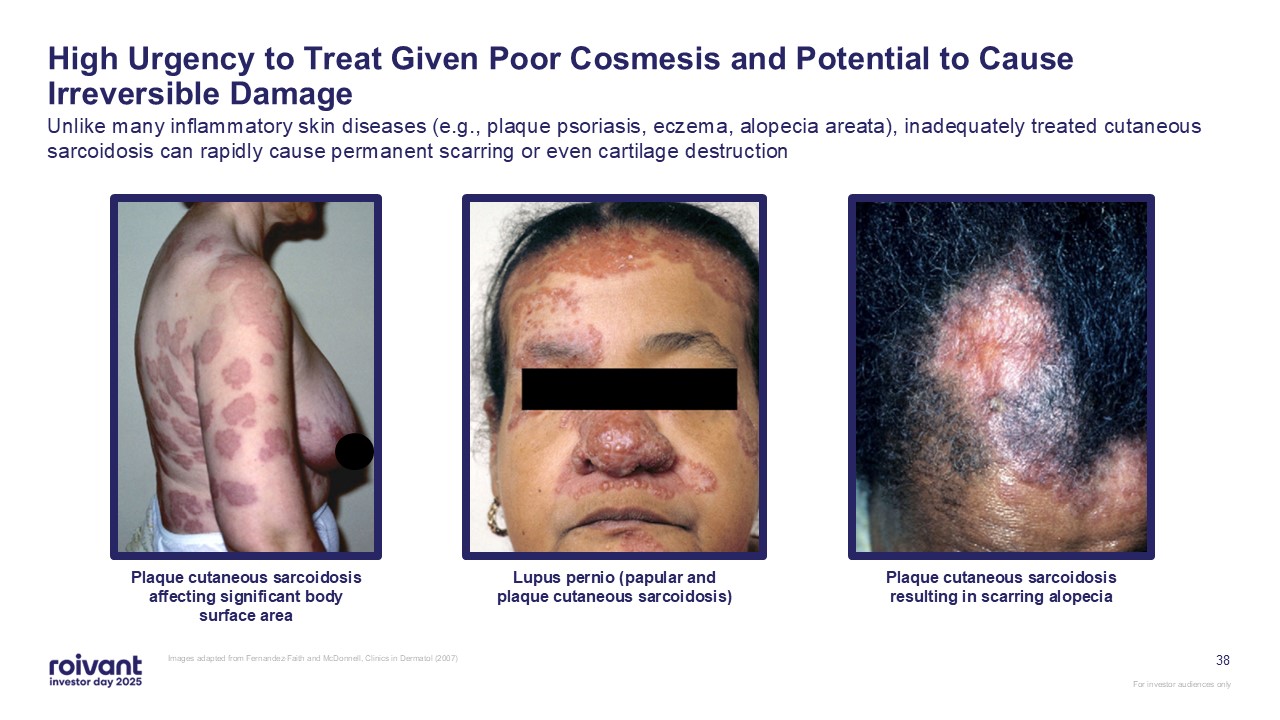
38 High Urgency to Treat Given Poor Cosmesis and Potential to Cause
Irreversible Damage Unlike many inflammatory skin diseases (e.g., plaque psoriasis, eczema, alopecia areata), inadequately treated cutaneous sarcoidosis can rapidly cause permanent scarring or even cartilage destruction Images adapted from
Fernandez-Faith and McDonnell, Clinics in Dermatol (2007) Plaque cutaneous sarcoidosis affecting significant body surface area Lupus pernio (papular and plaque cutaneous sarcoidosis) Plaque cutaneous sarcoidosis resulting in scarring
alopecia
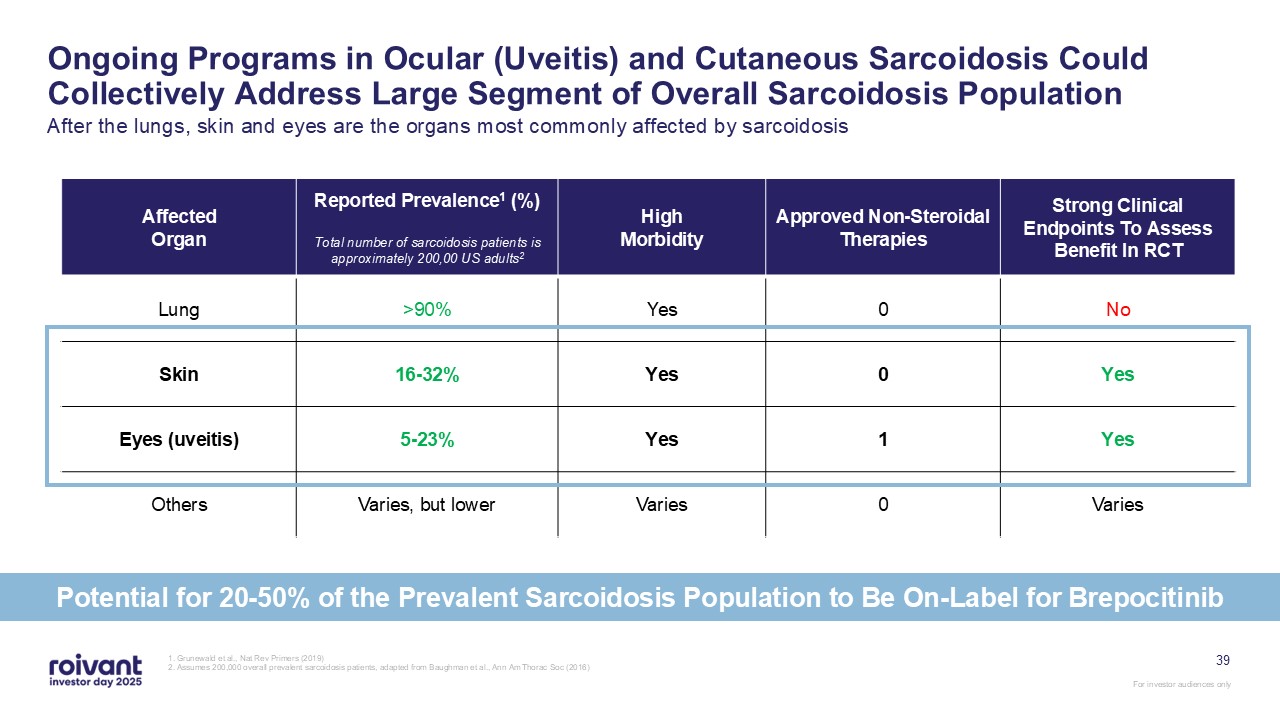
39 Ongoing Programs in Ocular (Uveitis) and Cutaneous Sarcoidosis Could
Collectively Address Large Segment of Overall Sarcoidosis Population After the lungs, skin and eyes are the organs most commonly affected by sarcoidosis 1. Grunewald et al., Nat Rev Primers (2019) 2. Assumes 200,000 overall prevalent
sarcoidosis patients, adapted from Baughman et al., Ann Am Thorac Soc (2016) Affected Organ Reported Prevalence1 (%) Total number of sarcoidosis patients is approximately 200,00 US adults2 High Morbidity Approved Non-Steroidal
Therapies Strong Clinical Endpoints To Assess Benefit In RCT Lung >90% Yes 0 No Skin 16-32% Yes 0 Yes Eyes (uveitis) 5-23% Yes 1 Yes Others Varies, but lower Varies 0 Varies Potential for 20-50% of the Prevalent
Sarcoidosis Population to Be On-Label for Brepocitinib

40 Cutaneous Sarcoidosis Alone Includes Eligible Population of ~40,000
Patients 1. Foundation for Sarcoidosis Research 2. Haimovic, et al., J Am Acad Dermatol (2012) 3. Altmeyer et al., Altmeyers Encycl (2025) ~40K CS patients in the US1,2 ~16K have minimal to no activity in other organs ~24K have
other organ involvement affecting treatment decisions (principally lung and/or eye)3 No approved therapies for pulmonary sarcoidosis, and none in late-stage clinical development Brepocitinib Has the Potential to Become the On-Label Therapy
of Choice for All Sarcoidosis Patients With Any Cutaneous Disease
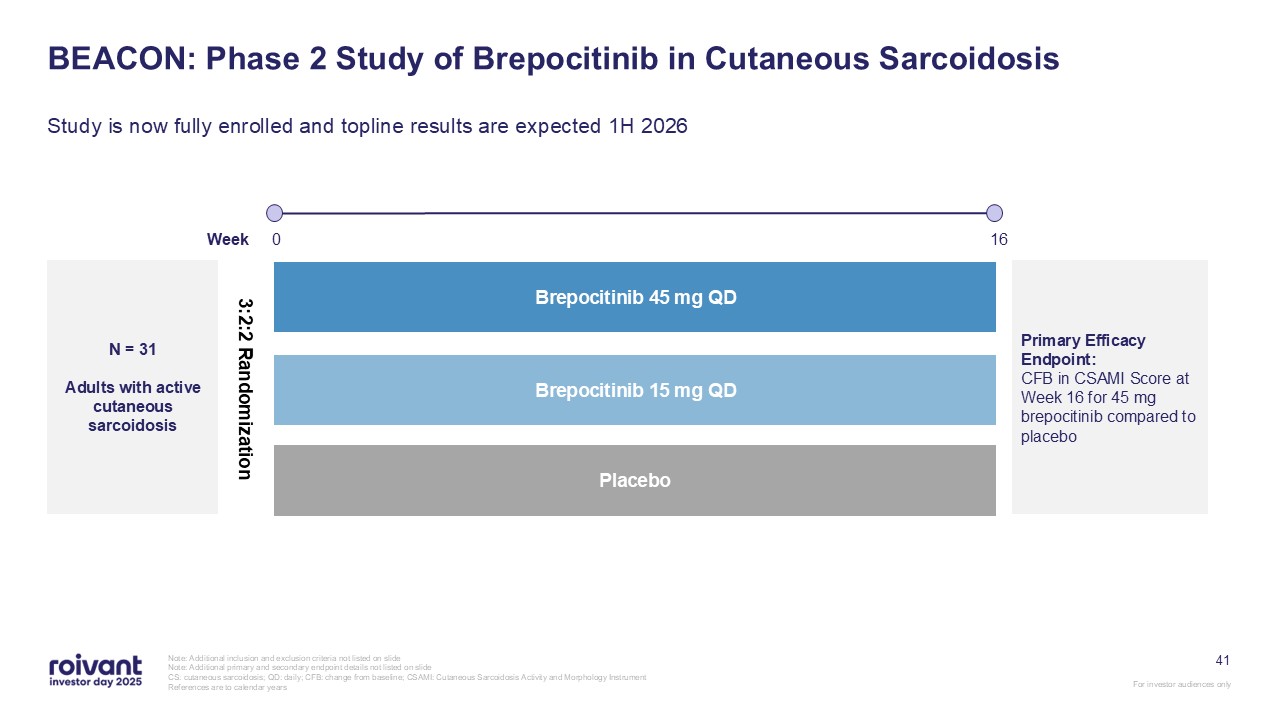
41 BEACON: Phase 2 Study of Brepocitinib in Cutaneous Sarcoidosis Study is now
fully enrolled and topline results are expected 1H 2026 Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide CS: cutaneous sarcoidosis; QD: daily;
CFB: change from baseline; CSAMI: Cutaneous Sarcoidosis Activity and Morphology Instrument References are to calendar years N = 31 Adults with active cutaneous sarcoidosis 16 Placebo Brepocitinib 45 mg QD Brepocitinib 15 mg
QD Week 0 Primary Efficacy Endpoint: CFB in CSAMI Score at Week 16 for 45 mg brepocitinib compared to placebo 3:2:2 Randomization

42 1. Noe et al., JAMA Dermatol (2020) BEACON’s Primary Efficacy Endpoint:
Change from Baseline in Cutaneous Sarcoidosis Activity & Morphology Index (CSAMI) CSAMI is analogous to other area-and-severity instruments CSAMI Activity (CSAMI-A) scores range from 0 – 165 Minimal clinically important difference
(MCID) = 5 pts1 Bar for Success in BEACON: At Least 5-Point Difference in Mean CSAMI-A CFB Between Brepocitinib 45 mg and Placebo, Supported by Totality of Patient-Level Data Across Endpoints

Dermatomyositis

Dermatomyositis Patient Video
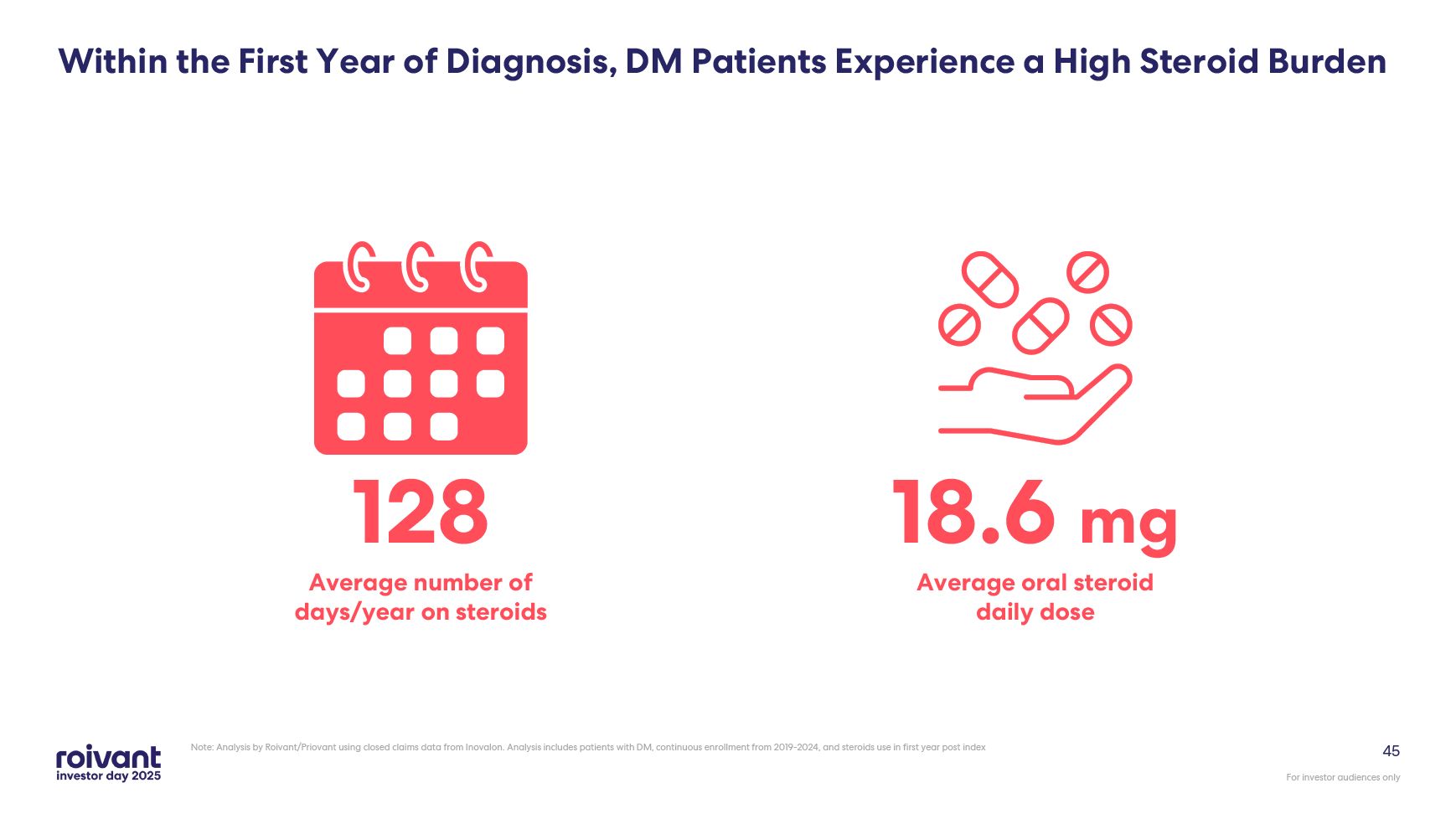
45 Within the First Year of Diagnosis, DM Patients Experience a High Steroid
Burden Note: Analysis by Roivant/Priovant using closed claims data from Inovalon. Analysis includes patients with DM, continuous enrollment from 2019-2024, and steroids use in first year post index 128 Average number of days/year on
steroids 18.6 mg Average oral steroid daily dose
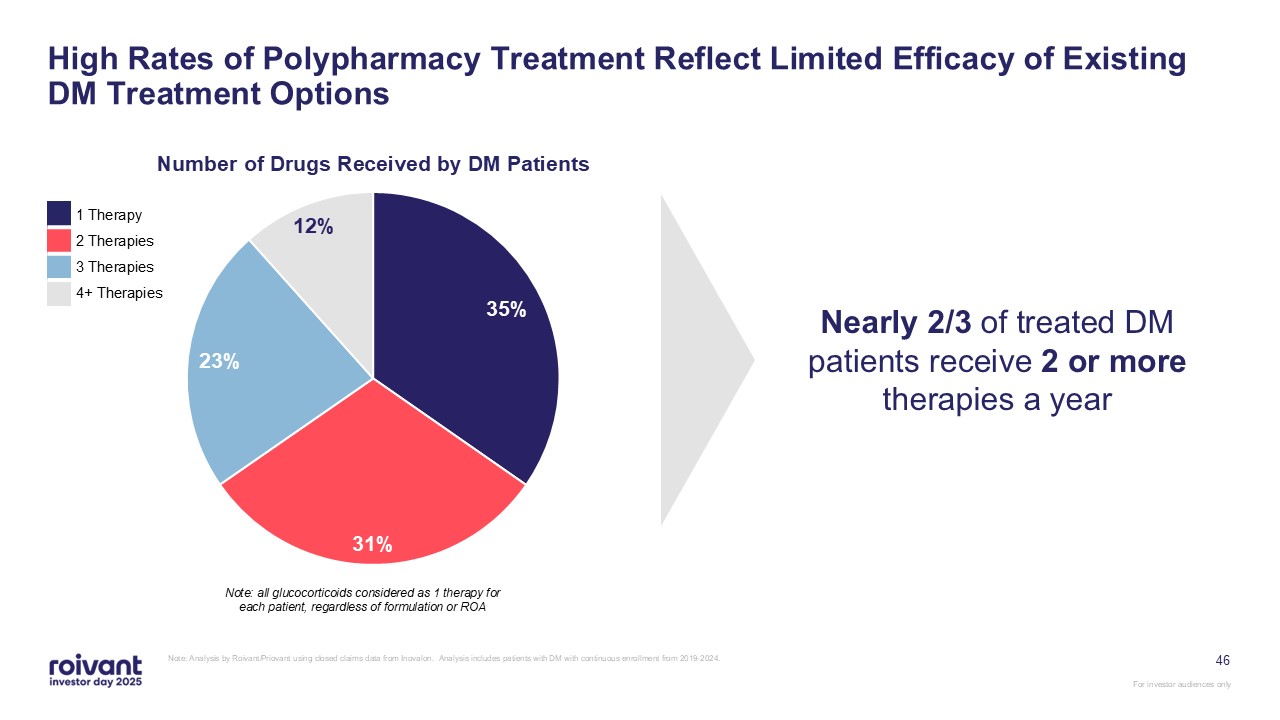
46 High Rates of Polypharmacy Treatment Reflect Limited Efficacy of Existing DM
Treatment Options Note: Analysis by Roivant/Priovant using closed claims data from Inovalon. Analysis includes patients with DM with continuous enrollment from 2019-2024. Note: all glucocorticoids considered as 1 therapy for each patient,
regardless of formulation or ROA Nearly 2/3 of treated DM patients receive 2 or more therapies a year 1 Therapy 2 Therapies 3 Therapies 4+ Therapies
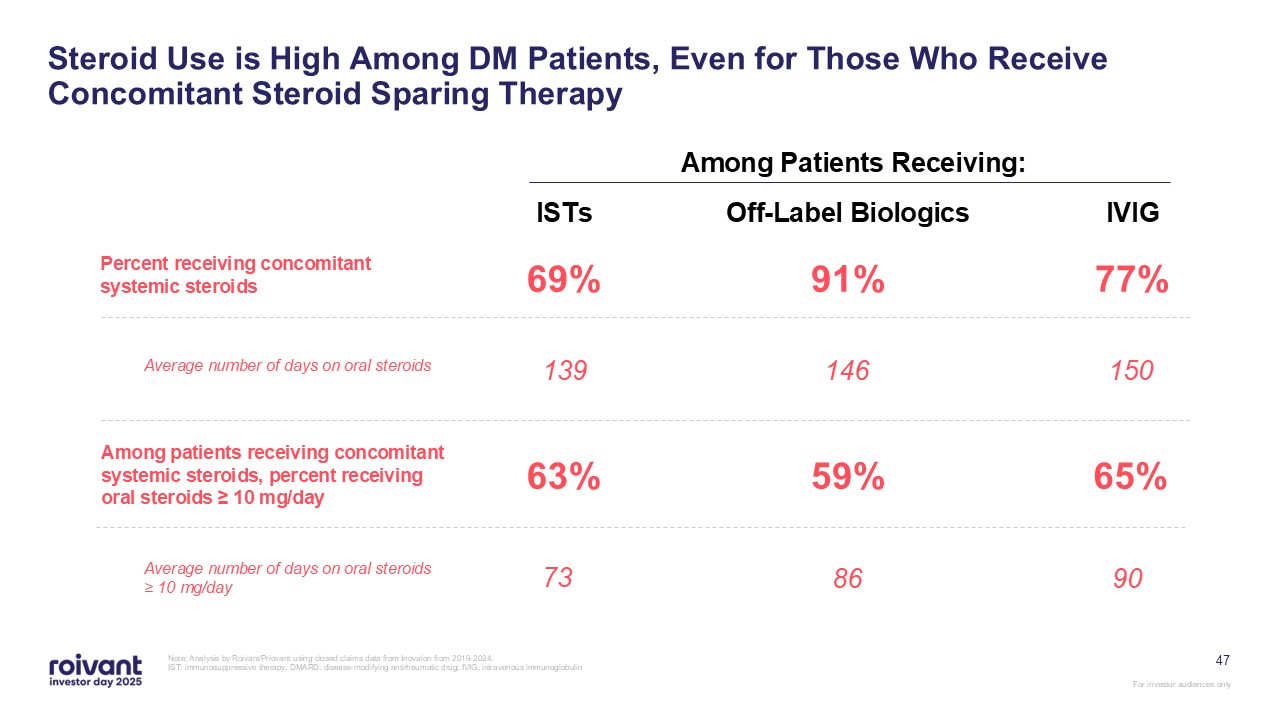
47 Steroid Use is High Among DM Patients, Even for Those Who Receive
Concomitant Steroid Sparing Therapy Note: Analysis by Roivant/Priovant using closed claims data from Inovalon from 2019-2024. IST: immunosuppressive therapy; DMARD: disease-modifying antirheumatic drug; IVIG: intravenous
immunoglobulin Among Patients Receiving: ISTs 69% 139 63% Off-Label Biologics 91% 146 59% IVIG 77% 150 65% Percent receiving concomitant systemic steroids Average number of days on oral steroids Among patients receiving
concomitant systemic steroids, percent receiving oral steroids ≥ 10 mg/day Average number of days on oral steroids ≥ 10 mg/day 73 86 90
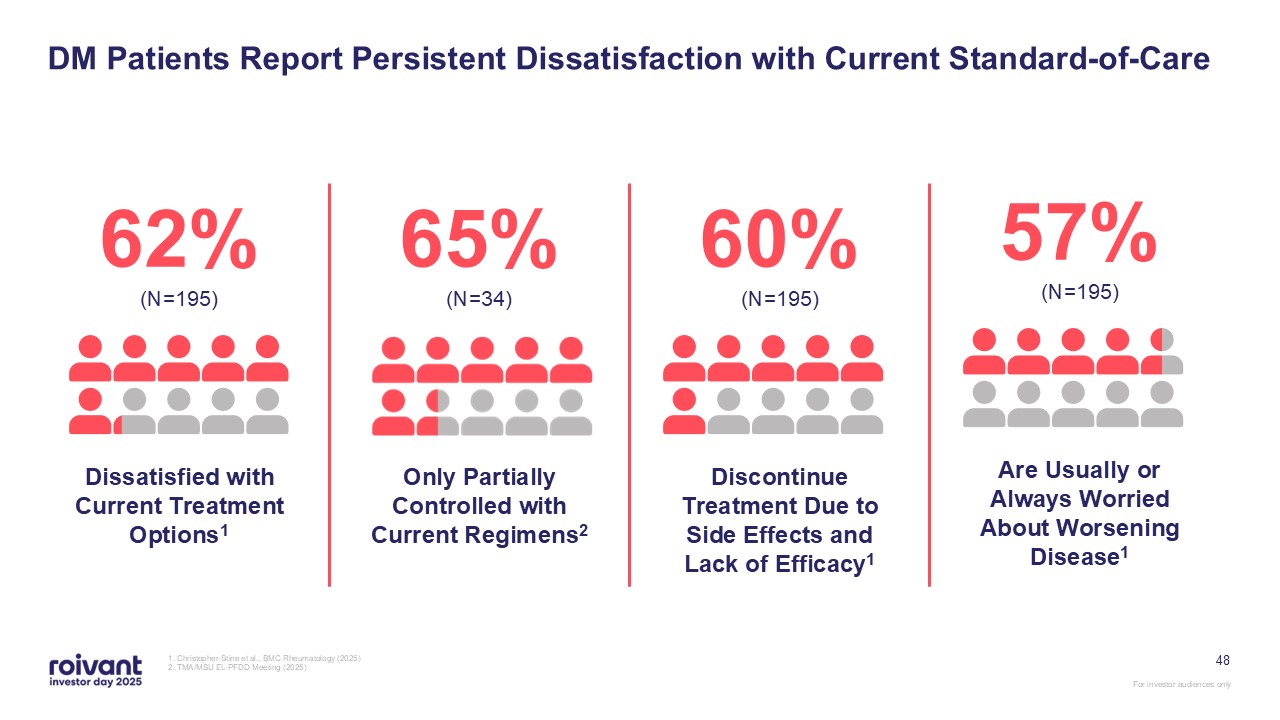
48 DM Patients Report Persistent Dissatisfaction with Current
Standard-of-Care 1. Christopher-Stine et al., BMC Rheumatology (2025) 2. TMA/MSU EL-PFDD Meeting (2025) 62% (N=195) Dissatisfied with Current Treatment Options1 65% (N=34) Only Partially Controlled with Current
Regimens2 60% (N=195) Discontinue Treatment Due to Side Effects and Lack of Efficacy1 57% (N=195) Are Usually or Always Worried About Worsening Disease1
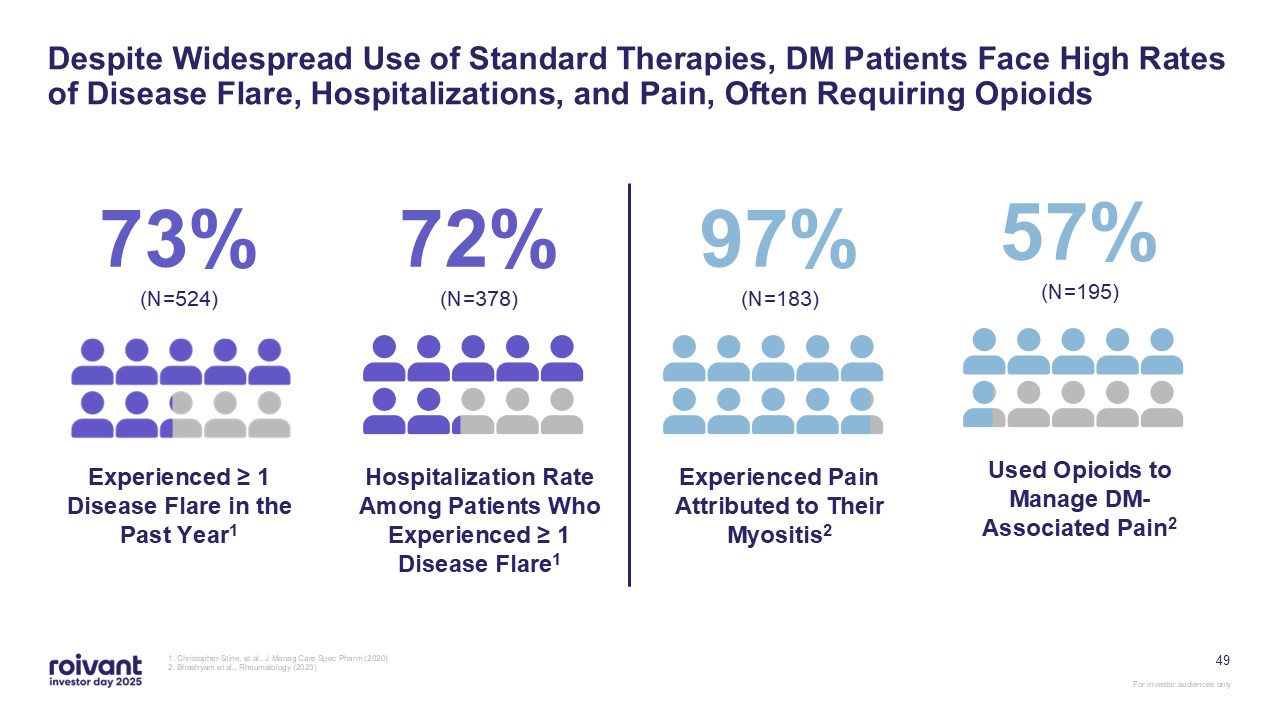
73% (N=524) Experienced ≥ 1 Disease Flare in the Past
Year1 72% (N=378) Hospitalization Rate Among Patients Who Experienced ≥ 1 Disease Flare1 97% (N=183) Experienced Pain Attributed to Their Myositis2 57% (N=195) Used Opioids to Manage DM-Associated Pain2 49 Despite Widespread Use
of Standard Therapies, DM Patients Face High Rates of Disease Flare, Hospitalizations, and Pain, Often Requiring Opioids 1. Christopher-Stine, et al., J Manag Care Spec Pharm (2020) 2. Bhashyam et al., Rheumatology (2023)

50 Patient Advocacy Group Surveys (TMA, MSU) Report Significant Muscle Disease
Burden and Impact on Patients’ ADLs 1. Priovant/TMA Patient Survey 2. Myositis Journey and Burden of Disease Survey, MSU (2022) 53%2 Unable to walk more than 1 mile Unable to lift or carry groceries 36%2 63%1 Unable to climb one
flight of stairs Unable to do chores (e.g., vacuuming, yard work) 40%2 50%2 Unable to bend, kneel, or stoop Report use of mobility aids (e.g., canes/crutches, rollators, walkers, and wheelchairs) 35%2
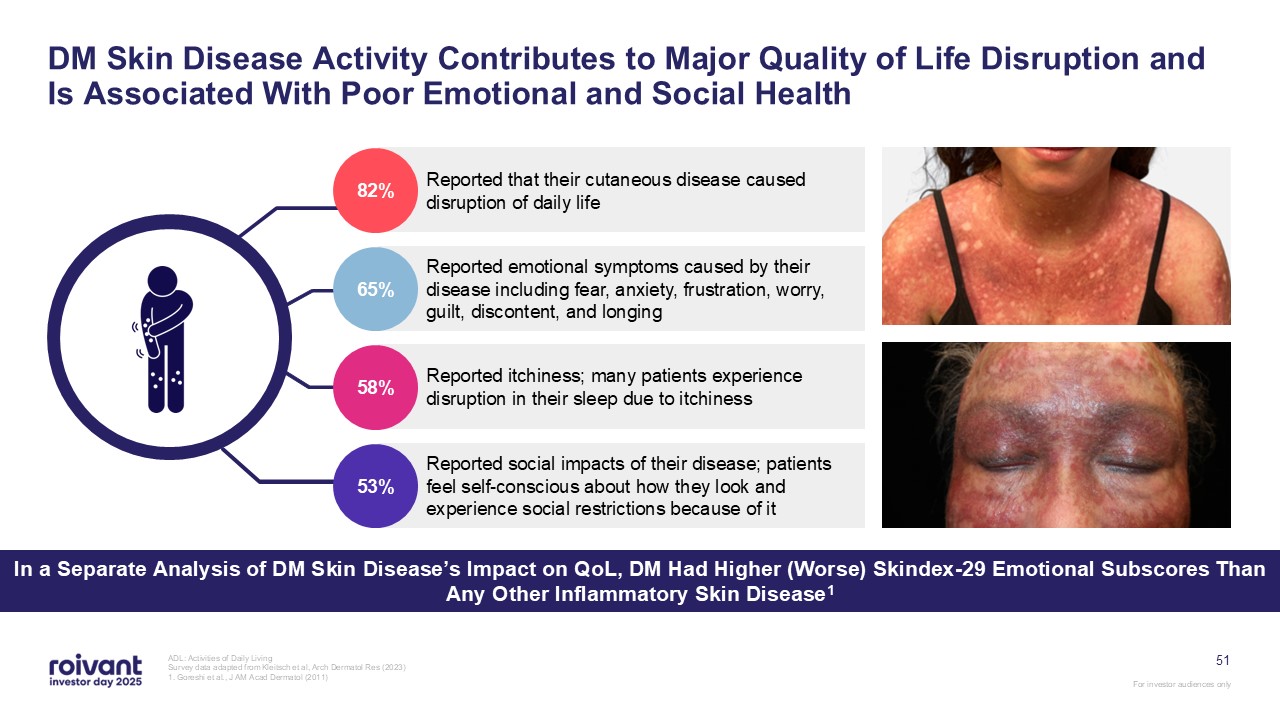
51 DM Skin Disease Activity Contributes to Major Quality of Life Disruption and
Is Associated With Poor Emotional and Social Health ADL: Activities of Daily Living Survey data adapted from Kleitsch et al, Arch Dermatol Res (2023) 1. Goreshi et al., J AM Acad Dermatol (2011) Reported that their cutaneous disease
caused disruption of daily life Reported emotional symptoms caused by their disease including fear, anxiety, frustration, worry, guilt, discontent, and longing Reported itchiness; many patients experience disruption in their sleep due to
itchiness Reported social impacts of their disease; patients feel self-conscious about how they look and experience social restrictions because of it 58% 53% 65% 82% In a Separate Analysis of DM Skin Disease’s Impact on QoL, DM Had
Higher (Worse) Skindex-29 Emotional Subscores Than Any Other Inflammatory Skin Disease1
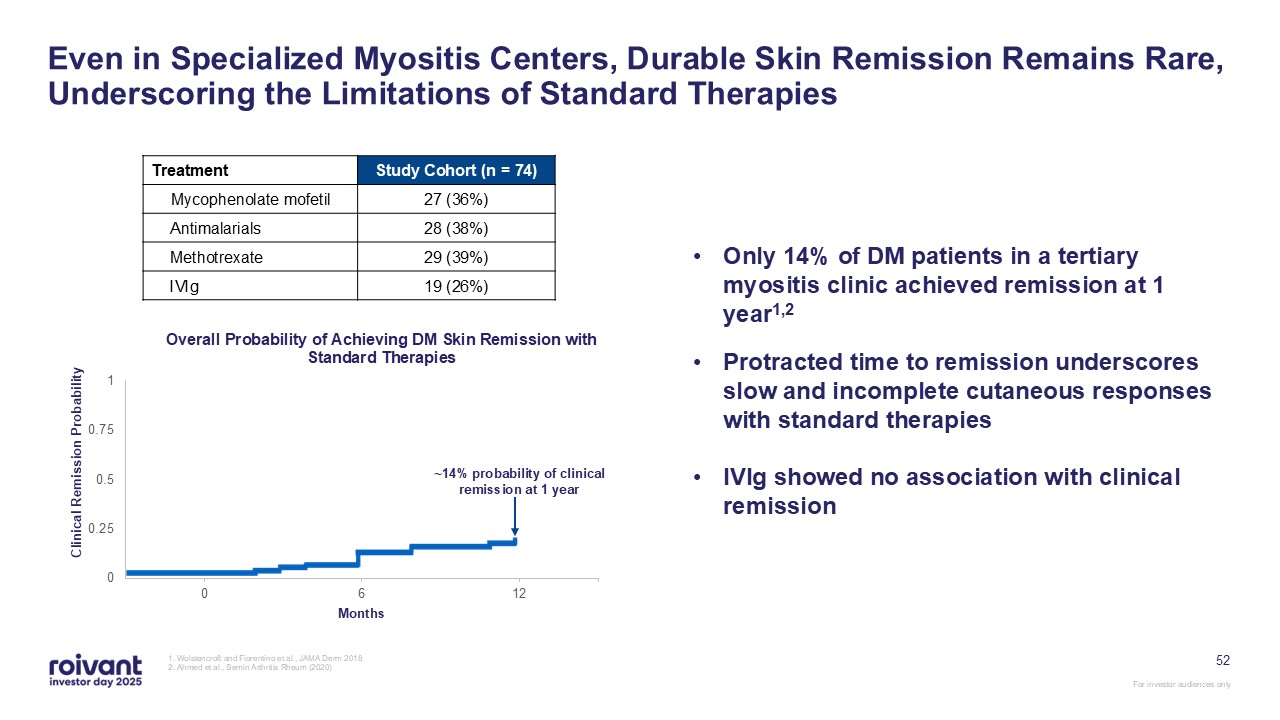
52 Even in Specialized Myositis Centers, Durable Skin Remission Remains Rare,
Underscoring the Limitations of Standard Therapies 1. Wolstencroft and Fiorentino et al., JAMA Derm 2018 2. Ahmed et al., Semin Arthritis Rheum (2020) Treatment Study Cohort (n = 74) Mycophenolate mofetil 27 (36%) Antimalarials 28
(38%) Methotrexate 29 (39%) IVIg 19 (26%) ~14% probability of clinical remission at 1 year Only 14% of DM patients in a tertiary myositis clinic achieved remission at 1 year1,2 Protracted time to remission underscores slow and
incomplete cutaneous responses with standard therapies IVIg showed no association with clinical remission
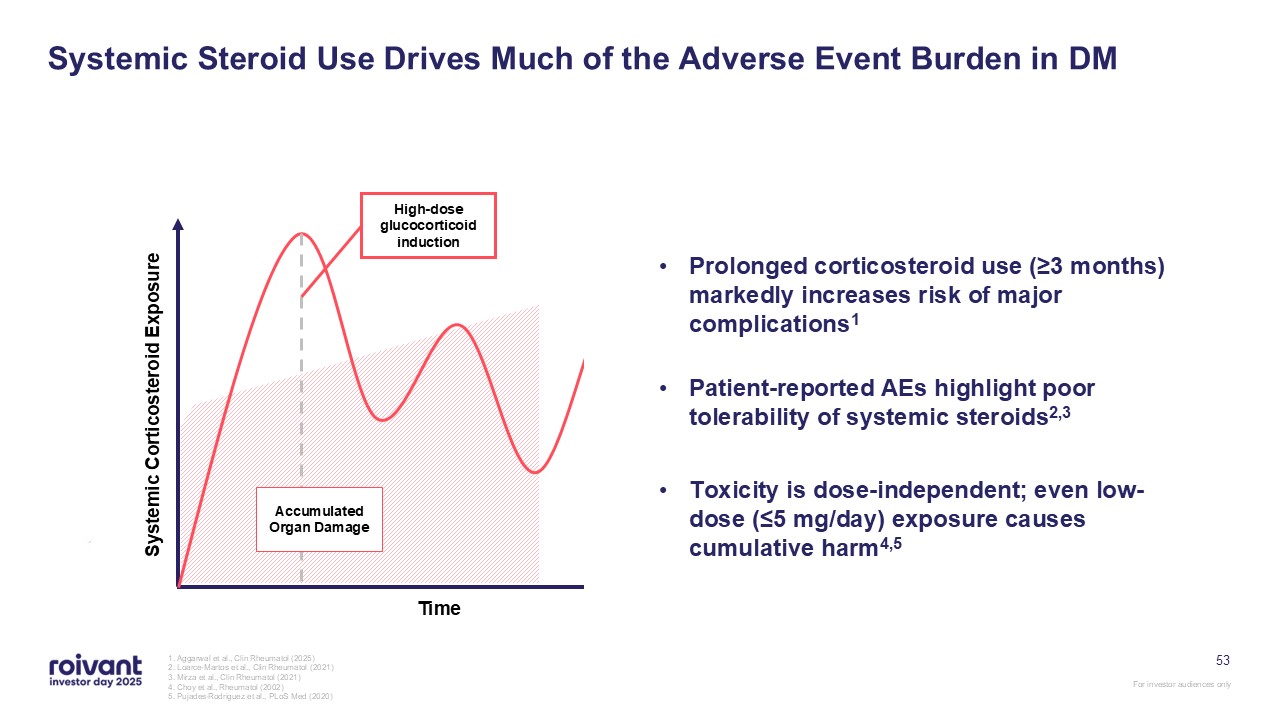
53 Systemic Steroid Use Drives Much of the Adverse Event Burden in DM 1.
Aggarwal et al., Clin Rheumatol (2025) 2. Loarce-Martos et al., Clin Rheumatol (2021) 3. Mirza et al., Clin Rheumatol (2021) 4. Choy et al., Rheumatol (2002) 5. Pujades-Rodriguez et al., PLoS Med (2020) Systemic Corticosteroid
Exposure Time High-dose glucocorticoid induction High-dose glucocorticoid induction Accumulated Organ Damage Prolonged corticosteroid use (≥3 months) markedly increases risk of major complications1 Patient-reported AEs highlight poor
tolerability of systemic steroids2,3 Toxicity is dose-independent; even low-dose (≤5 mg/day) exposure causes cumulative harm4,5
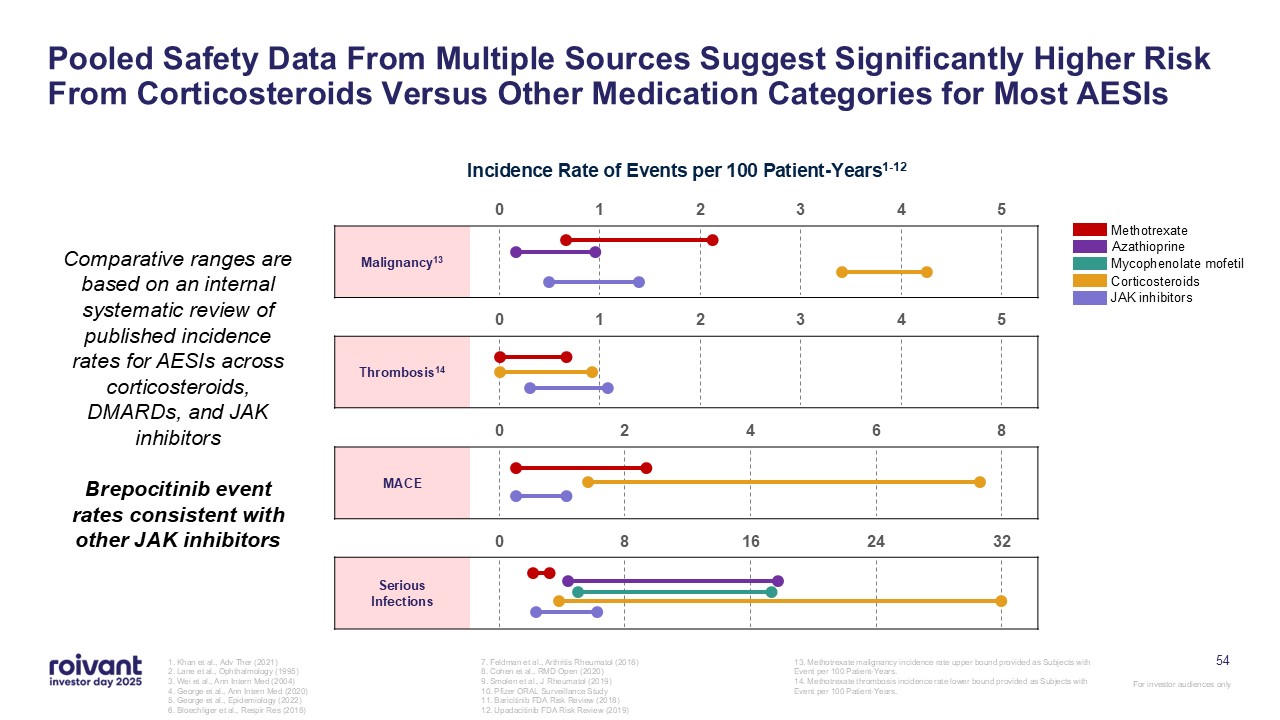
54 Pooled Safety Data From Multiple Sources Suggest Significantly Higher Risk
From Corticosteroids Versus Other Medication Categories for Most AESIs 1. Khan et al., Adv Ther (2021) 2. Lane et al., Ophthalmology (1995) 3. Wei et al., Ann Intern Med (2004) 4. George et al., Ann Intern Med (2020) 5. George et al.,
Epidemiology (2022) 6. Bloechliger et al., Respir Res (2018) 7. Feldman et al., Arthritis Rheumatol (2018) 8. Cohen et al., RMD Open (2020) 9. Smolen et al., J Rheumatol (2019) 10. Pfizer ORAL Surveillance Study 11. Baricitinib FDA Risk
Review (2018) 12. Upadacitinib FDA Risk Review (2019) 13. Methotrexate malignancy incidence rate upper bound provided as Subjects with Event per 100 Patient-Years. 14. Methotrexate thrombosis incidence rate lower bound provided as Subjects
with Event per 100 Patient-Years. Serious Infections MACE Thrombosis14 Malignancy13 Methotrexate Azathioprine Mycophenolate mofetil Corticosteroids JAK inhibitors Incidence Rate of Events per 100 Patient-Years1-12 Comparative
ranges are based on an internal systematic review of published incidence rates for AESIs across corticosteroids, DMARDs, and JAK inhibitors Brepocitinib event rates consistent with other JAK inhibitors
Patients are heavily treated with polypharmacy, including high dose OCS
administered chronically1 Patient are unhappy with the current treatment options and are frequently switching their treatment2 Patients report continued symptoms, flares, and pain despite treatment3,4 Continued symptoms are leading to
significant burden on ADLs, QoL, and overall health outcomes5,6 These adverse health outcomes are compounded by the toxicities of high-dose chronic steroids7-9 High unmet need for novel, targeted therapy that can provide sustained clinical
benefit while allowing patients to get to minimal or no steroid burden 55 DM Patient Experience Shows Need for New Treatments That Can Meaningfully Impact Patients’ Quality of Life 1. Analysis by Roivant/Priovant using closed claims data
from Inovalon from 2019-2024 2. Christopher-Stine et al., BMC Rheumatology (2025) 3. Christopher-Stine, et al., J Manag Care Spec Pharm (2020) 4. Bhashyam et al., Rheumatology (2023) 5. Kleitsch et al., Arch Dermatol Res (2023) 6.
Goreshi et al., J AM Acad Dermatol (2011) 7. Aggarwal et al., Clin Rheumatol (2025) 8. Choy et al., Rheumatol (2002) 9. Pujades-Rodriguez et al., PLoS Med (2020)
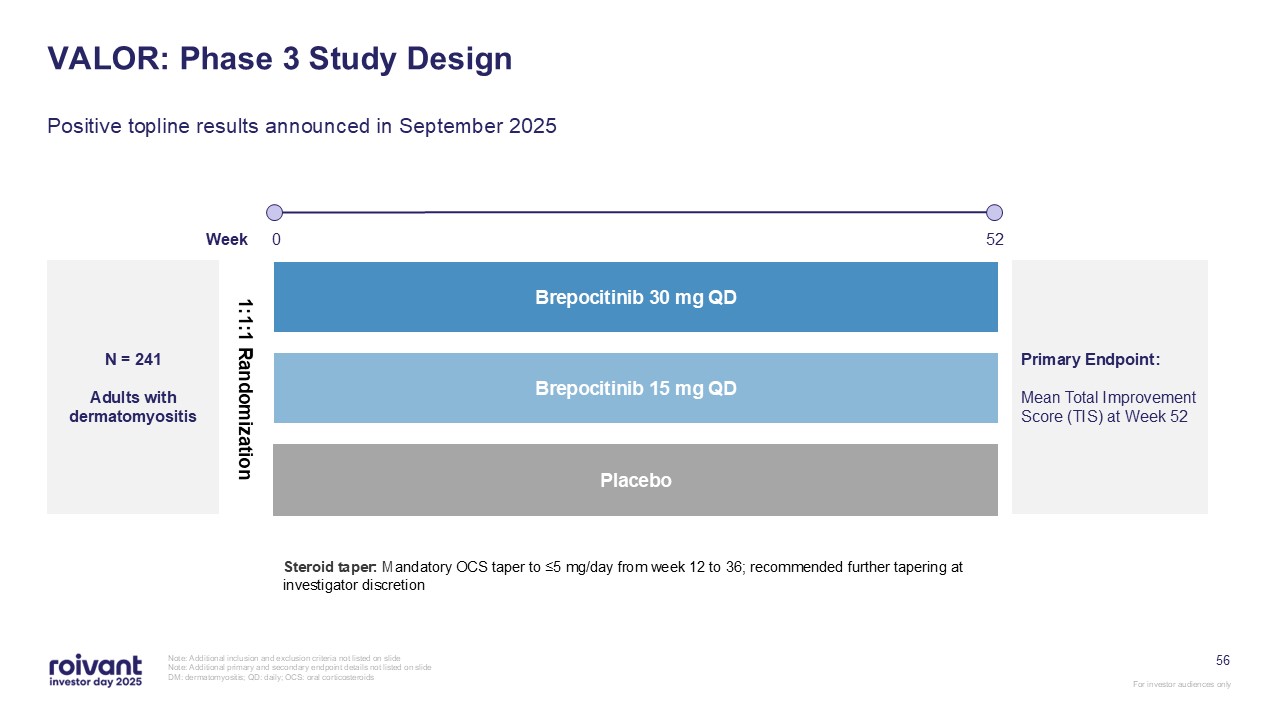
56 VALOR: Phase 3 Study Design Positive topline results announced in September
2025 Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide DM: dermatomyositis; QD: daily; OCS: oral corticosteroids N = 241 Adults with
dermatomyositis 1:1:1 Randomization Placebo Brepocitinib 30 mg QD Brepocitinib 15 mg QD Week 0 52 Primary Endpoint: Mean Total Improvement Score (TIS) at Week 52 Steroid taper: Mandatory OCS taper to ≤5 mg/day from week 12 to 36;
recommended further tapering at investigator discretion
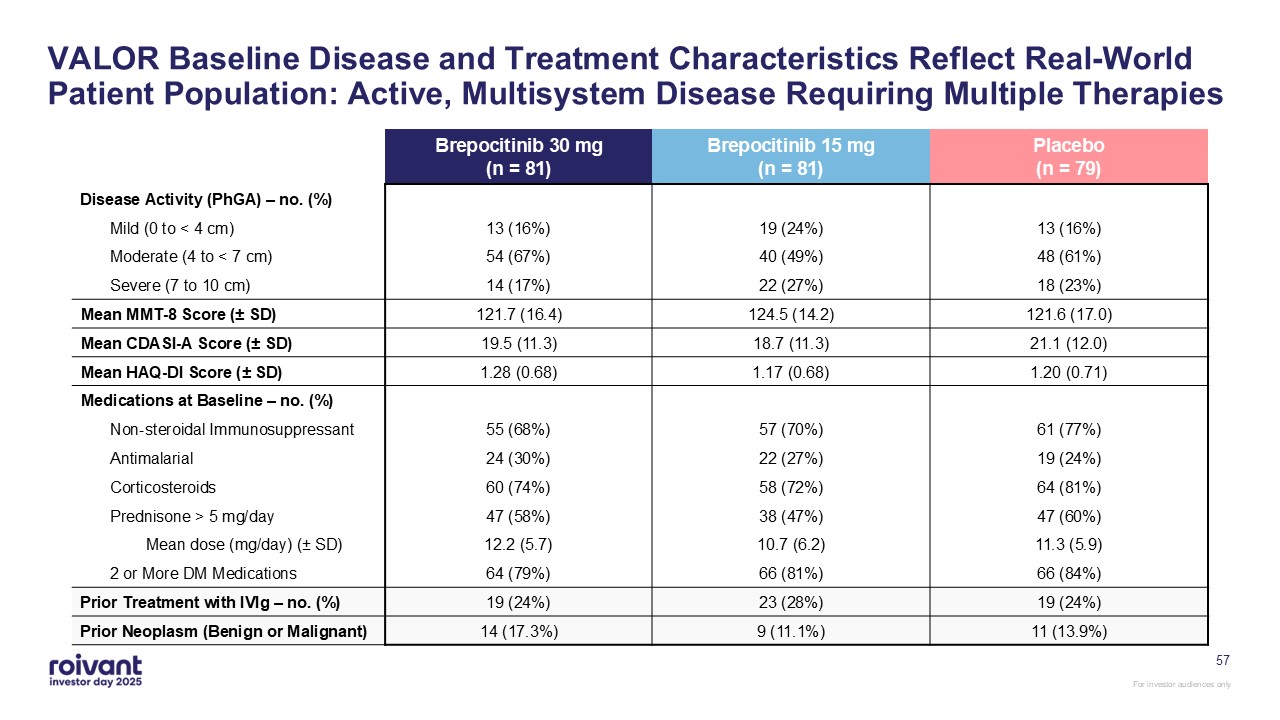
57 VALOR Baseline Disease and Treatment Characteristics Reflect Real-World
Patient Population: Active, Multisystem Disease Requiring Multiple Therapies Brepocitinib 30 mg(n = 81) Brepocitinib 15 mg(n = 81) Placebo(n = 79) Disease Activity (PhGA) – no. (%) Mild (0 to < 4 cm) 13 (16%) 19 (24%) 13
(16%) Moderate (4 to < 7 cm) 54 (67%) 40 (49%) 48 (61%) Severe (7 to 10 cm) 14 (17%) 22 (27%) 18 (23%) Mean MMT-8 Score (± SD) 121.7 (16.4) 124.5 (14.2) 121.6 (17.0) Mean CDASI-A Score (± SD) 19.5 (11.3) 18.7 (11.3) 21.1
(12.0) Mean HAQ-DI Score (± SD) 1.28 (0.68) 1.17 (0.68) 1.20 (0.71) Medications at Baseline – no. (%) Non-steroidal Immunosuppressant 55 (68%) 57 (70%) 61 (77%) Antimalarial 24 (30%) 22 (27%) 19 (24%) Corticosteroids 60
(74%) 58 (72%) 64 (81%) Prednisone > 5 mg/day 47 (58%) 38 (47%) 47 (60%) Mean dose (mg/day) (± SD) 12.2 (5.7) 10.7 (6.2) 11.3 (5.9) 2 or More DM Medications 64 (79%) 66 (81%) 66 (84%) Prior Treatment with IVIg – no. (%) 19
(24%) 23 (28%) 19 (24%) Prior Neoplasm (Benign or Malignant) 14 (17.3%) 9 (11.1%) 11 (13.9%)

Brepocitinib 30 mg Achieved Statistically Significant Benefit On All Ten Ranked
Endpoints in the VALOR Study Measurements of skin disease, muscle disease, rapidity of onset, and steroid sparing; consistent dose response was also seen across endpoints Key Endpoint Important Features Brepocitinib 30mg
(n=81) Placebo (n=79) P-Value Mean TIS (Primary) Composite endpoint, focus on muscle disease and global benefit 46.5 31.2 0.0006 CDASI-A change from baseline at Week 52 Improvement in skin disease
activity -11.7 -7.0 0.0006 DMOMS at Week 52 DM-specific muscle and skin composite measure of benefit 57.9 40.5 0.0014 TIS40 Response at Week 52 Moderate TIS response (focus on global benefit / muscle) 67.9% 44.3% 0.0040 Time to
Consecutive TIS40 Response by Week 52 Time to onset of sustained benefit (particularly high bar) 85 days 168 days 0.0155 Patients achieving TIS40 Response + ≤2.5 mg OCS at Week 52 Achievement of clinical response and steroid
reduction 54.3% 26.6% 0.0006 CDASI-A 40% Response with ≥4-point improvement at Week 52 Clinically meaningful skin response 61.7% 44.3% 0.0357 TIS60 Response at Week 52 Major TIS response – Highest TIS response
threshold 46.1% 26.4% 0.0126 Change from baseline in HAQ-DI at Week 52 Improvement in physical and functional disability and daily living activities related to muscle strength -0.337 -0.042 0.0035 Change from baseline in CDASI-A at
Week 4 Rapid onset of skin response -6.4 -3.5 0.0003 58
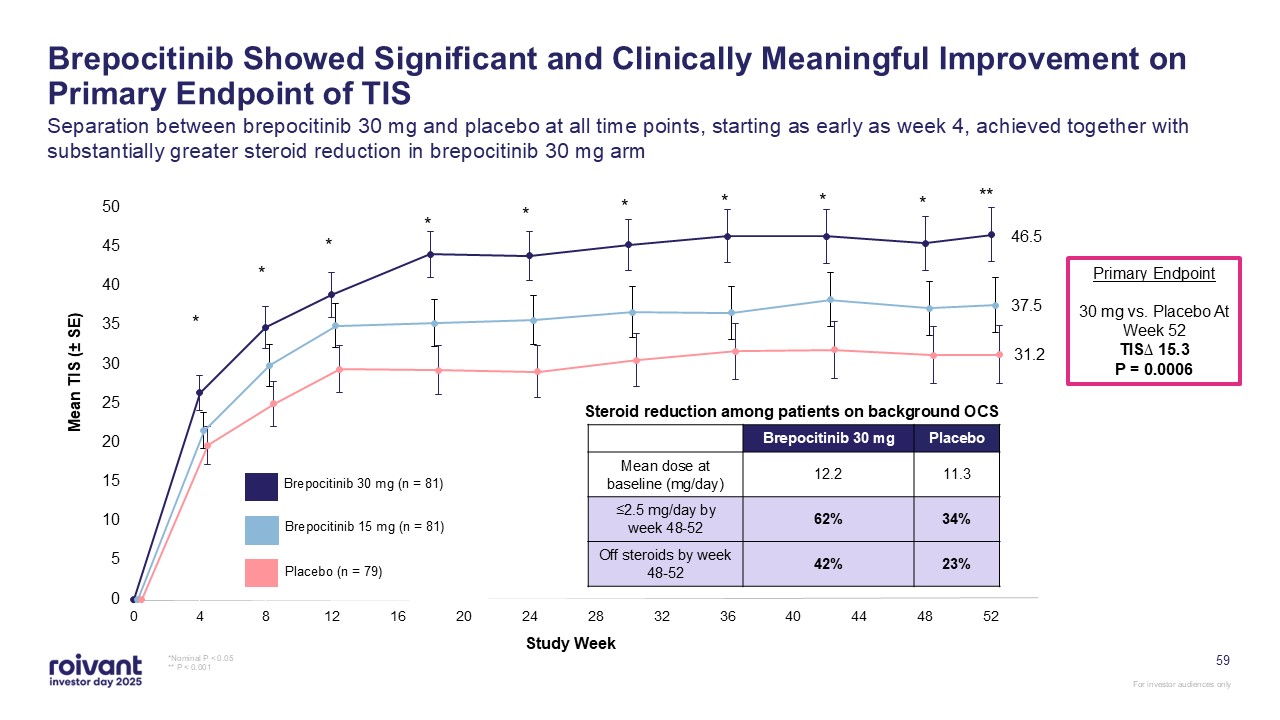
Brepocitinib Showed Significant and Clinically Meaningful Improvement on Primary
Endpoint of TIS Separation between brepocitinib 30 mg and placebo at all time points, starting as early as week 4, achieved together with substantially greater steroid reduction in brepocitinib 30 mg arm *Nominal P < 0.05 ** P <
0.001 Brepocitinib 30 mg (n = 81) Brepocitinib 15 mg (n = 81) Placebo (n = 79) Study Week * * * * * * * * * ** 46.5 37.5 31.2 Primary Endpoint 30 mg vs. Placebo At Week 52 TIS∆ 15.3 P = 0.0006 Brepocitinib 30
mg Placebo Mean dose at baseline (mg/day) 12.2 11.3 ≤2.5 mg/day by week 48-52 62% 34% Off steroids by week 48-52 42% 23% Steroid reduction among patients on background OCS 59
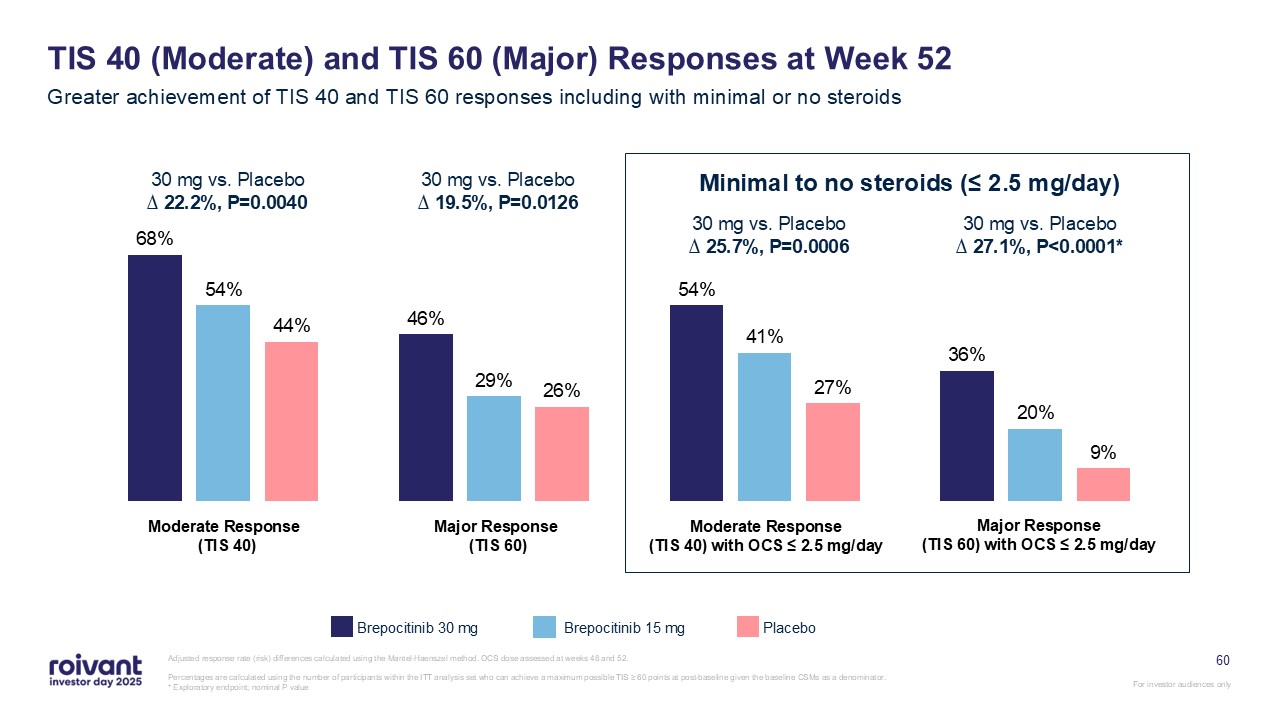
60 TIS 40 (Moderate) and TIS 60 (Major) Responses at Week 52 Greater
achievement of TIS 40 and TIS 60 responses including with minimal or no steroids Adjusted response rate (risk) differences calculated using the Mantel-Haenszel method. OCS dose assessed at weeks 48 and 52. Percentages are calculated using
the number of participants within the ITT analysis set who can achieve a maximum possible TIS ≥ 60 points at post-baseline given the baseline CSMs as a denominator.* Exploratory endpoint; nominal P value Brepocitinib 30 mg Brepocitinib 15
mg Placebo 30 mg vs. Placebo∆ 22.2%, P=0.0040 30 mg vs. Placebo∆ 19.5%, P=0.0126 30 mg vs. Placebo∆ 25.7%, P=0.0006 30 mg vs. Placebo∆ 27.1%, P<0.0001* Minimal to no steroids (≤ 2.5 mg/day) Moderate Response (TIS 40) Major Response
(TIS 60) Moderate Response (TIS 40) with OCS ≤ 2.5 mg/day Major Response (TIS 60) with OCS ≤ 2.5 mg/day
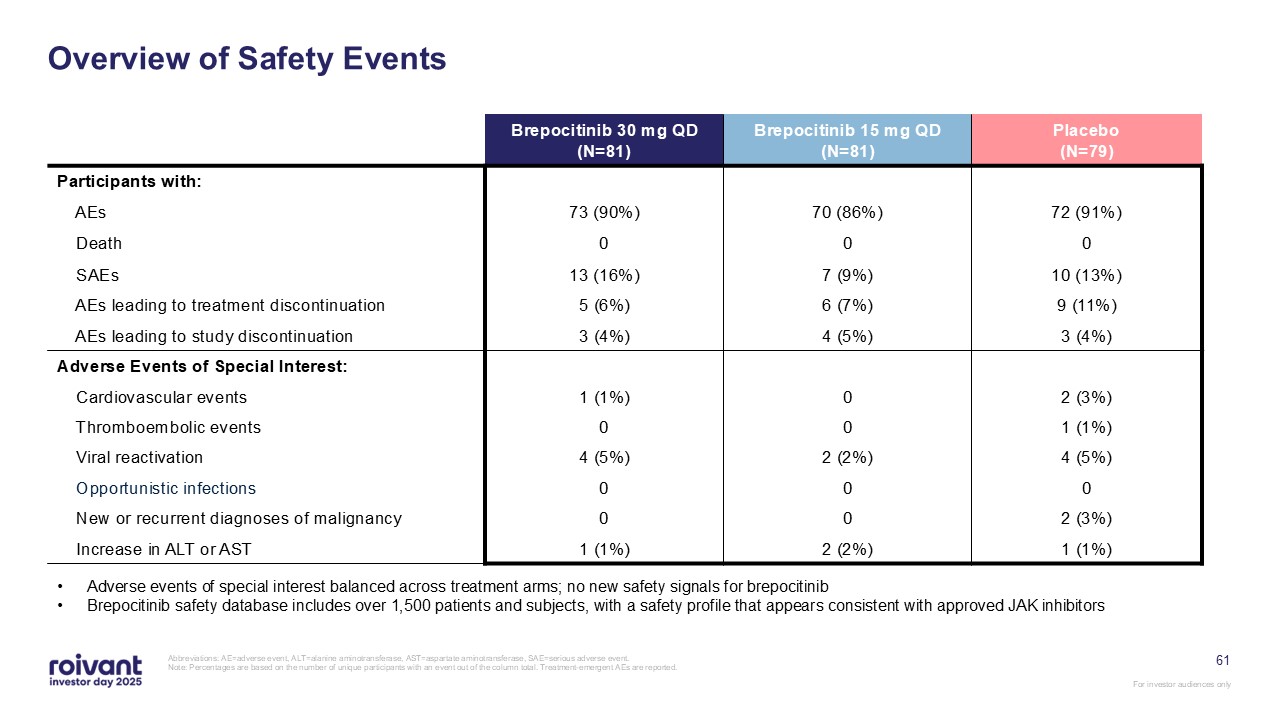
61 Overview of Safety Events Abbreviations: AE=adverse event, ALT=alanine
aminotransferase, AST=aspartate aminotransferase, SAE=serious adverse event. Note: Percentages are based on the number of unique participants with an event out of the column total. Treatment-emergent AEs are reported. Brepocitinib 30 mg
QD(N=81) Brepocitinib 15 mg QD(N=81) Placebo(N=79) Participants with: AEs 73 (90%) 70 (86%) 72 (91%) Death 0 0 0 SAEs 13 (16%) 7 (9%) 10 (13%) AEs leading to treatment discontinuation 5 (6%) 6 (7%) 9 (11%) AEs leading
to study discontinuation 3 (4%) 4 (5%) 3 (4%) Adverse Events of Special Interest: Cardiovascular events 1 (1%) 0 2 (3%) Thromboembolic events 0 0 1 (1%) Viral reactivation 4 (5%) 2 (2%) 4 (5%) Opportunistic
infections 0 0 0 New or recurrent diagnoses of malignancy 0 0 2 (3%) Increase in ALT or AST 1 (1%) 2 (2%) 1 (1%) Adverse events of special interest balanced across treatment arms; no new safety signals for
brepocitinib Brepocitinib safety database includes over 1,500 patients and subjects, with a safety profile that appears consistent with approved JAK inhibitors

Key Regulatory and Launch Planning Activities Strong engagement with patient
community Patient advocacy group collaborations and events Dermatomyositis.com disease website and associated social media ecosystem Limited distribution network and in-house Priovant Hub Strategy consistent with prior successful rare
disease launches Partner selection and operational buildout well underway Robust field medical team in place driving scientific engagement with key DM-treating physicians Foundational relationships from before VALOR TLR with at least one
key physician at all top DM centers of excellence in US Post-TLR engagement has expanded to include additional relevant HCPs at centers of excellence, as well as longer tail of community specialists NDA Submission Expected in Early
2026 62 Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All
references are to calendar years.
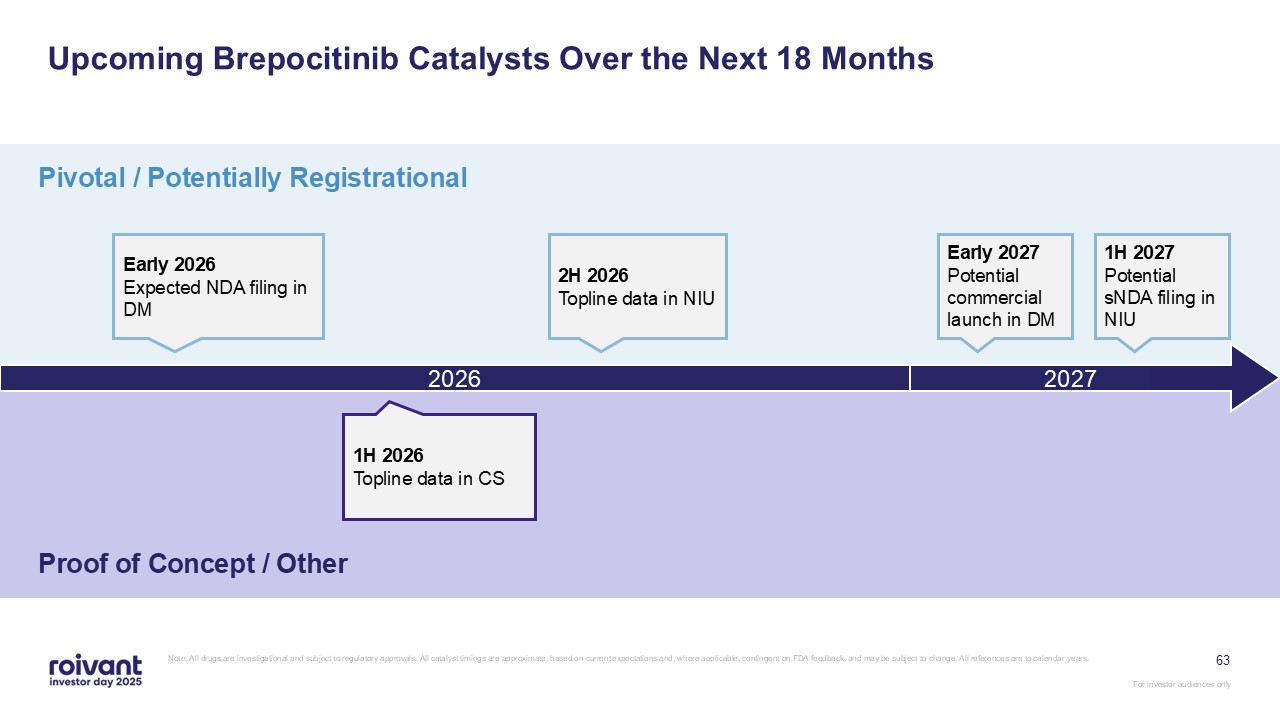
Upcoming Brepocitinib Catalysts Over the Next 18 Months Pivotal / Potentially
Registrational Proof of Concept / Other Early 2026 Expected NDA filing in DM 2026 2H 2026 Topline data in NIU 1H 2027 Potential sNDA filing in NIU 1H 2026 Topline data in CS 63 2027 Early 2027 Potential commercial launch in DM
Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are
to calendar years.

64 In Summary: Brepocitinib Phase 2 BEACON study for brepocitinib in CS
expected to read out 1H 2026; CS has high unmet medical need Following positive VALOR readout, planning for commercial launch of brepocitinib in DM is underway, with launch expected early 2027 Note: All drugs are investigational and subject
to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years. Phase 3 CLARITY study for
brepocitinib in NIU expected to read out 2H 2026; NIU has significant unmet medical need For investor audiences only

Q&A

66 IMVT-1402 Eric Venker CEO, Immunovant Matt Gline CEO, Roivant

67 IMVT-1402 drives deep dose-dependent reductions of pathogenic IgG
autoantibodies; expected to reach best-in-class IgG reductions of ~80%, unmatched by current anti-FcRn competitors Pipeline-in-a-product potential; approved anti-FcRns antibodies have generated ~$7BN in cumulative revenue in MG and CIDP
within 4 years of launch with additional indications expected1 Massive opportunity in uncontrolled Graves’ disease; generated disease-modifying PoC data and expect potentially registrational data in 2027 with multi-year lead and
best-in-class efficacy Significant evidence across late-stage clinical trials shows deeper IgG reductions are correlated with better efficacy across 8 different indications to date IMVT-1402 is expected to be first- and best-in-class in GD,
D2T RA, and CLE; best-in-class in MG, CIDP, and SjD; D2T RA topline readout now expected in 2026 as well as initial results in CLE Key Takeaways: IMVT-1402 Note: GD: Graves’ disease; SjD: Sjogren’s disease; D2T RA: difficult-to-treat
rheumatoid arthritis; CIDP: Chronic inflammatory demyelinating polyneuropathy; MG: Myasthenia gravis; CLE: Cutaneous lupus erythematosus Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are
approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years. 1. Data from Evaluate and company filings For investor audiences only
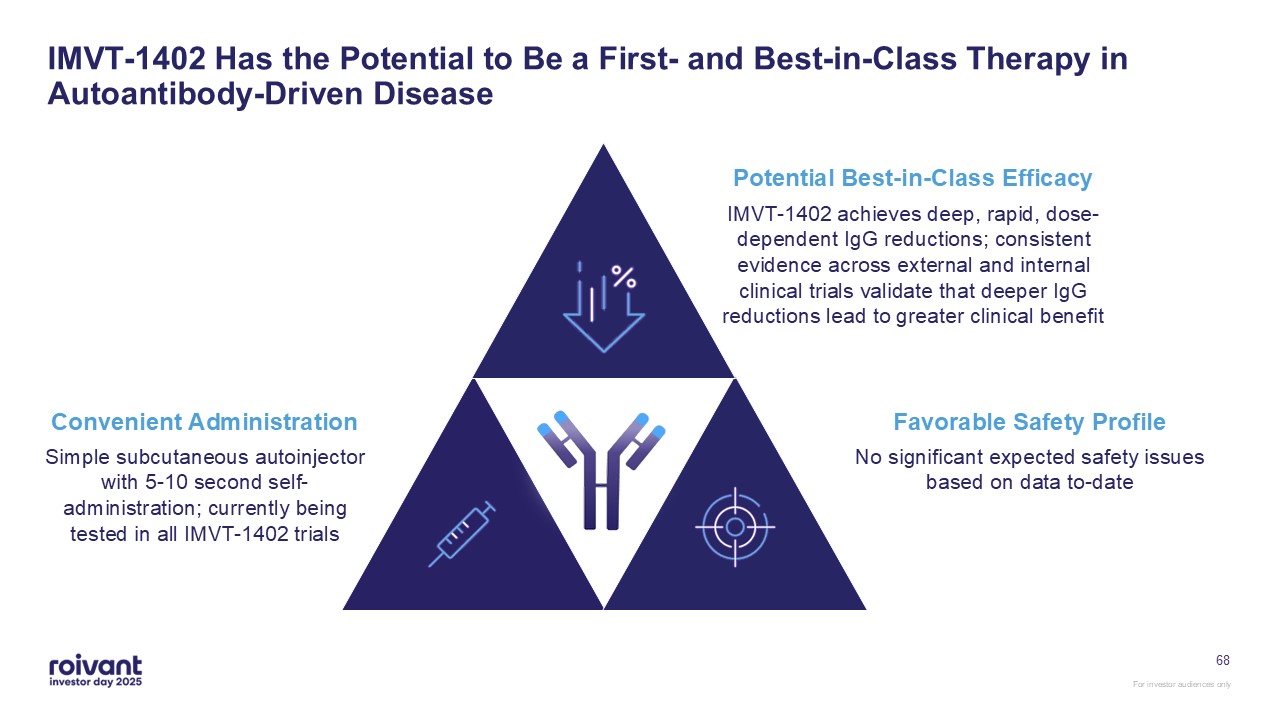
68 IMVT-1402 Has the Potential to Be a First- and Best-in-Class Therapy in
Autoantibody-Driven Disease Favorable Safety Profile Convenient Administration Simple subcutaneous autoinjector with 5-10 second self-administration; currently being tested in all IMVT-1402 trials Potential Best-in-Class
Efficacy IMVT-1402 achieves deep, rapid, dose-dependent IgG reductions; consistent evidence across external and internal clinical trials validate that deeper IgG reductions lead to greater clinical benefit No significant expected safety
issues based on data to-date
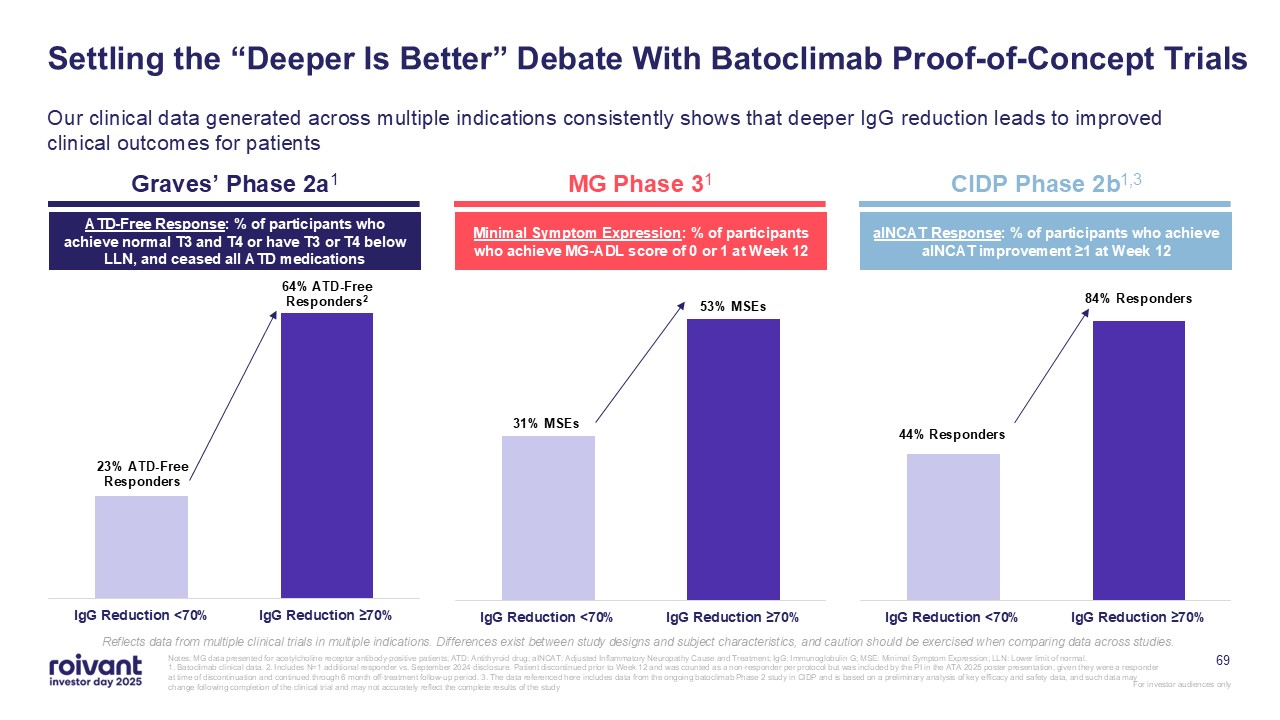
Settling the “Deeper Is Better” Debate With Batoclimab Proof-of-Concept
Trials Our clinical data generated across multiple indications consistently shows that deeper IgG reduction leads to improved clinical outcomes for patients Graves’ Phase 2a1 MG Phase 31 CIDP Phase 2b1,3 ATD-Free Response: % of
participants who achieve normal T3 and T4 or have T3 or T4 below LLN, and ceased all ATD medications Minimal Symptom Expression: % of participants who achieve MG-ADL score of 0 or 1 at Week 12 aINCAT Response: % of participants who achieve
aINCAT improvement ≥1 at Week 12 Reflects data from multiple clinical trials in multiple indications. Differences exist between study designs and subject characteristics, and caution should be exercised when comparing data across studies.
69 Notes: MG data presented for acetylcholine receptor antibody-positive patients; ATD: Antithyroid drug; aINCAT: Adjusted Inflammatory Neuropathy Cause and Treatment; IgG: Immunoglobulin G; MSE: Minimal Symptom Expression; LLN: Lower
limit of normal. 1. Batoclimab clinical data. 2. Includes N=1 additional responder vs. September 2024 disclosure. Patient discontinued prior to Week 12 and was counted as a non-responder per protocol but was included by the PI in the ATA
2025 poster presentation, given they were a responder at time of discontinuation and continued through 6 month off-treatment follow-up period. 3. The data referenced here includes data from the ongoing batoclimab Phase 2 study in CIDP and is
based on a preliminary analysis of key efficacy and safety data, and such data may change following completion of the clinical trial and may not accurately reflect the complete results of the study
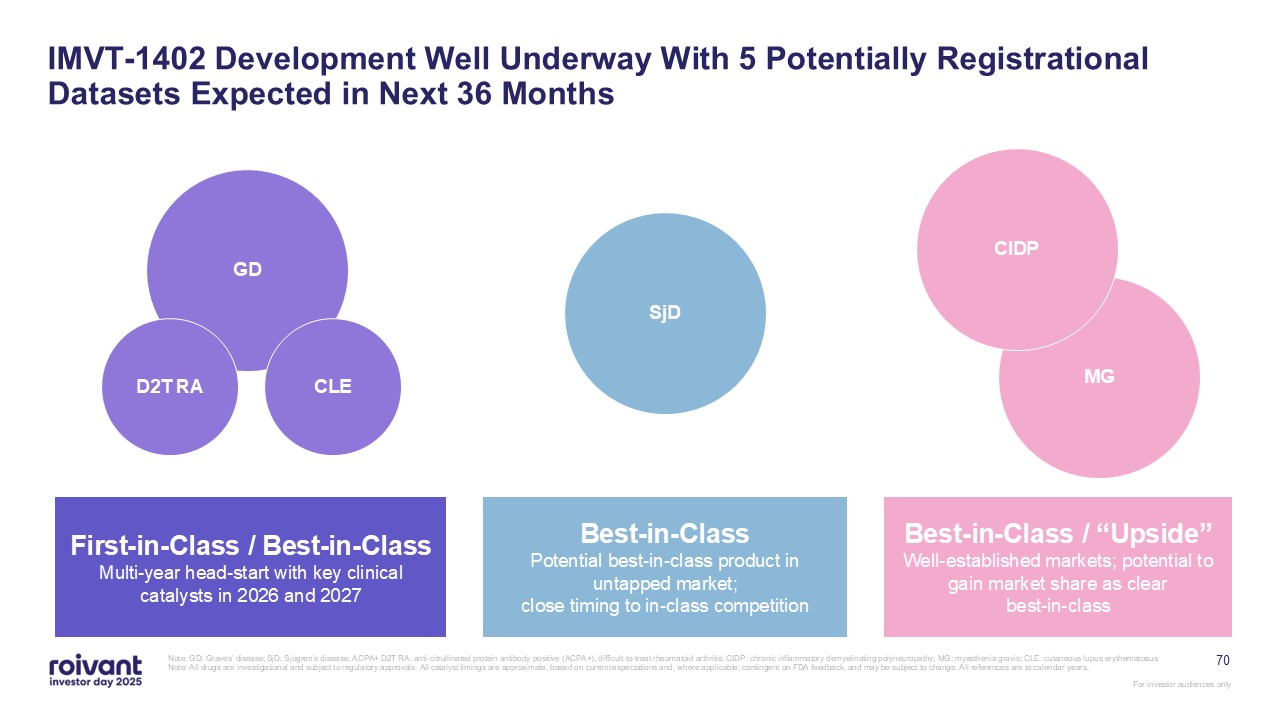
70 IMVT-1402 Development Well Underway With 5 Potentially Registrational
Datasets Expected in Next 36 Months Note: GD: Graves’ disease; SjD: Sjogren’s disease; ACPA+ D2T RA: anti-citrullinated protein antibody positive (ACPA+), difficult-to-treat rheumatoid arthritis; CIDP: chronic inflammatory demyelinating
polyneuropathy; MG: myasthenia gravis; CLE: cutaneous lupus erythematosus Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable,
contingent on FDA feedback, and may be subject to change. All references are to calendar years. MG CIDP SjD GD CLE D2T RA First-in-Class / Best-in-Class Multi-year head-start with key clinical catalysts in 2026 and
2027 Best-in-Class Potential best-in-class product in untapped market; close timing to in-class competition Best-in-Class / “Upside” Well-established markets; potential to gain market share as clear best-in-class
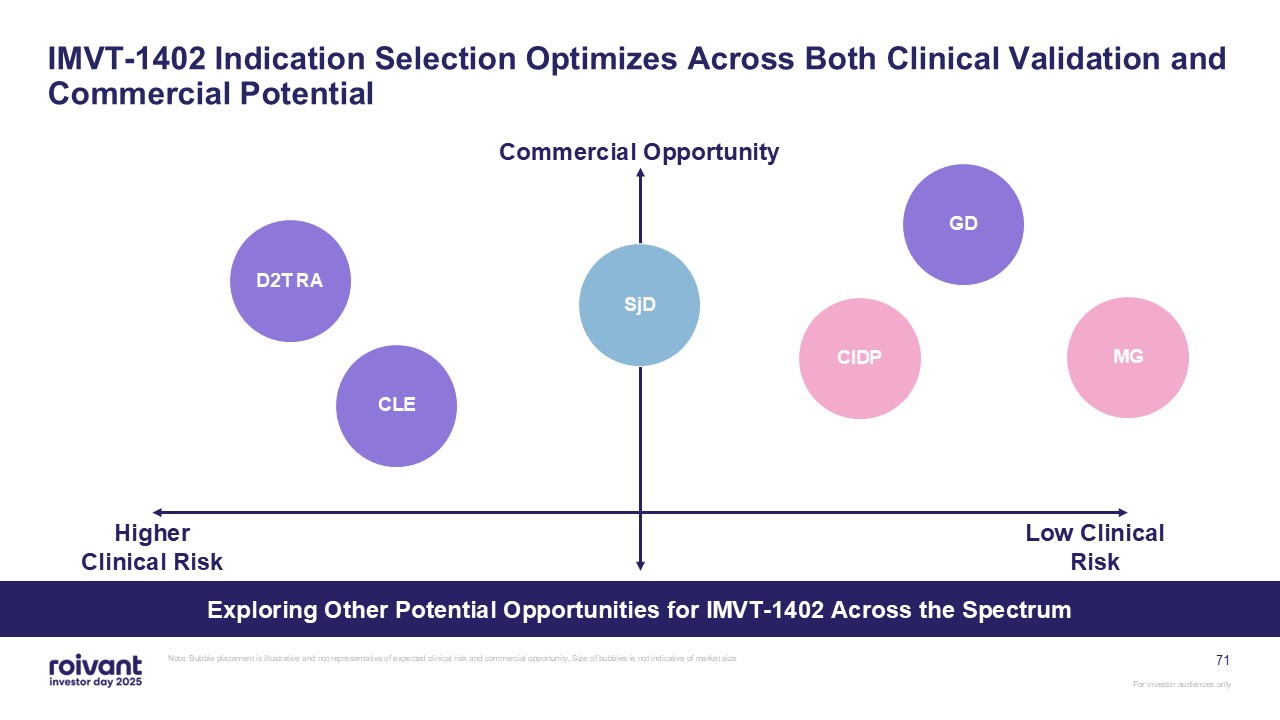
71 IMVT-1402 Indication Selection Optimizes Across Both Clinical Validation and
Commercial Potential Note: Bubble placement is illustrative and not representative of expected clinical risk and commercial opportunity. Size of bubbles is not indicative of market size Commercial Opportunity Low Clinical
Risk GD MG CIDP SjD D2T RA CLE Higher Clinical Risk Exploring Other Potential Opportunities for IMVT-1402 Across the Spectrum
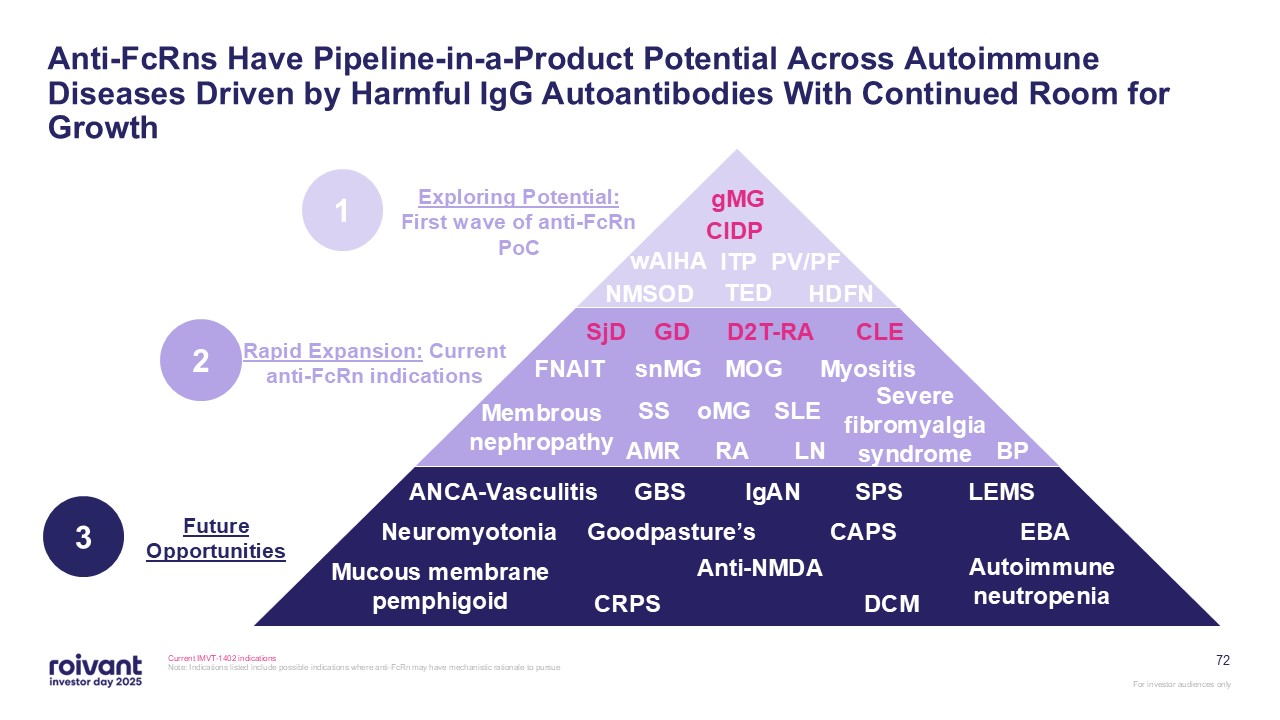
72 Anti-FcRns Have Pipeline-in-a-Product Potential Across Autoimmune Diseases
Driven by Harmful IgG Autoantibodies With Continued Room for Growth Current IMVT-1402 indications Note: Indications listed include possible indications where anti-FcRn may have mechanistic rationale to
pursue gMG CIDP wAIHA ITP PV/PF TED HDFN NMSOD MOG snMG GD oMG FNAIT D2T-RA Myositis SjD RA SLE Severe fibromyalgia
syndrome BP SS AMR LN CLE IgAN GBS CAPS SPS ANCA-Vasculitis Neuromyotonia Goodpasture’s CRPS Autoimmune neutropenia EBA Mucous membrane pemphigoid LEMS Anti-NMDA DCM Membrous nephropathy Exploring Potential: First wave of
anti-FcRn PoC Future Opportunities 1 Rapid Expansion: Current anti-FcRn indications 2 3
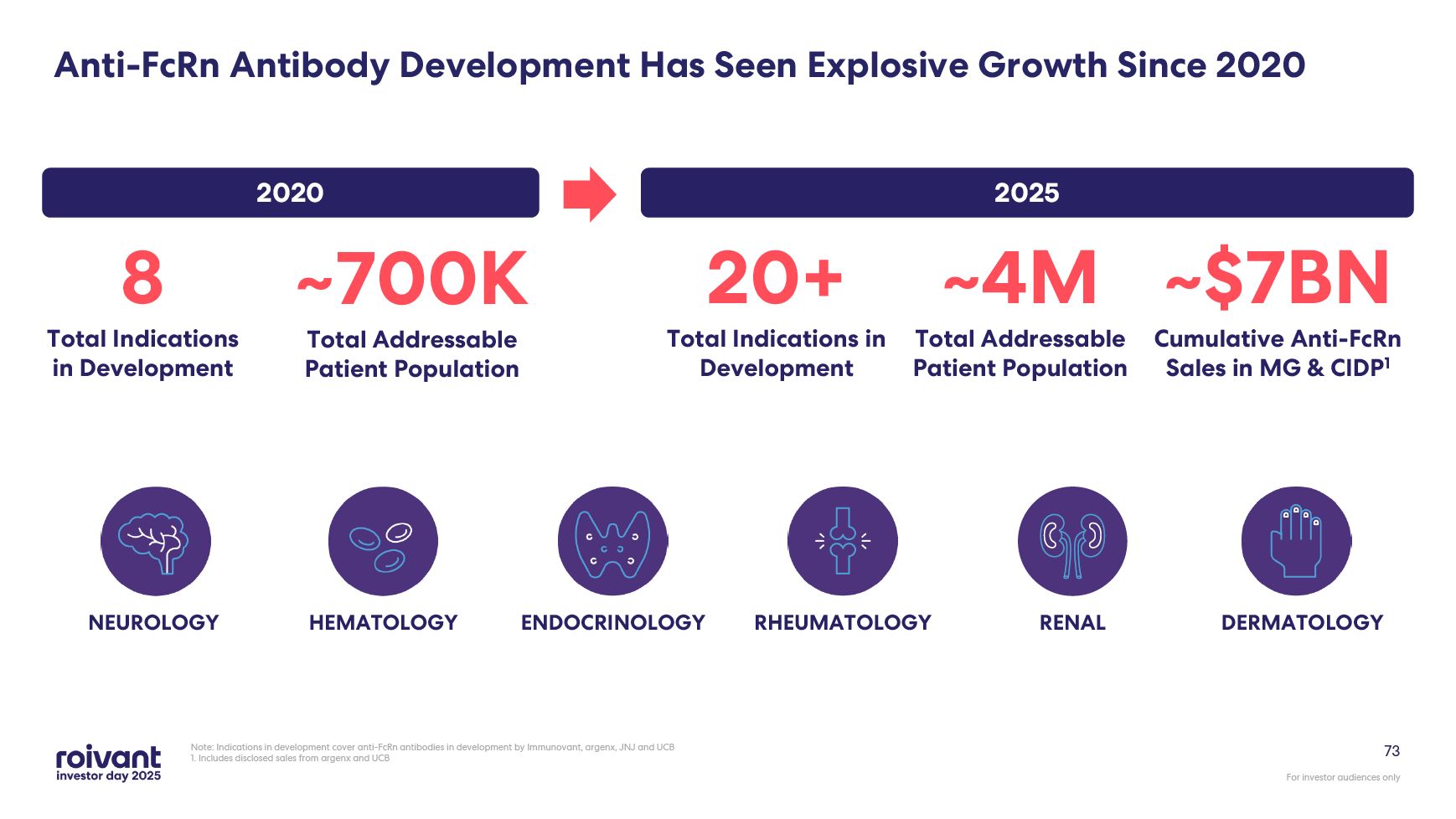
73 Note: Indications in development cover anti-FcRn antibodies in development
by Immunovant, argenx, JNJ and UCB 1. Includes disclosed sales from argenx and UCB ENDOCRINOLOGY RHEUMATOLOGY NEUROLOGY DERMATOLOGY RENAL HEMATOLOGY 8 Total Indications in Development ~700K Total Addressable Patient
Population 20+ Total Indications in Development ~4M Total Addressable Patient Population 2020 2025 Anti-FcRn Antibody Development Has Seen Explosive Growth Since 2020 ~$7BN Cumulative Anti-FcRn Sales in MG & CIDP1
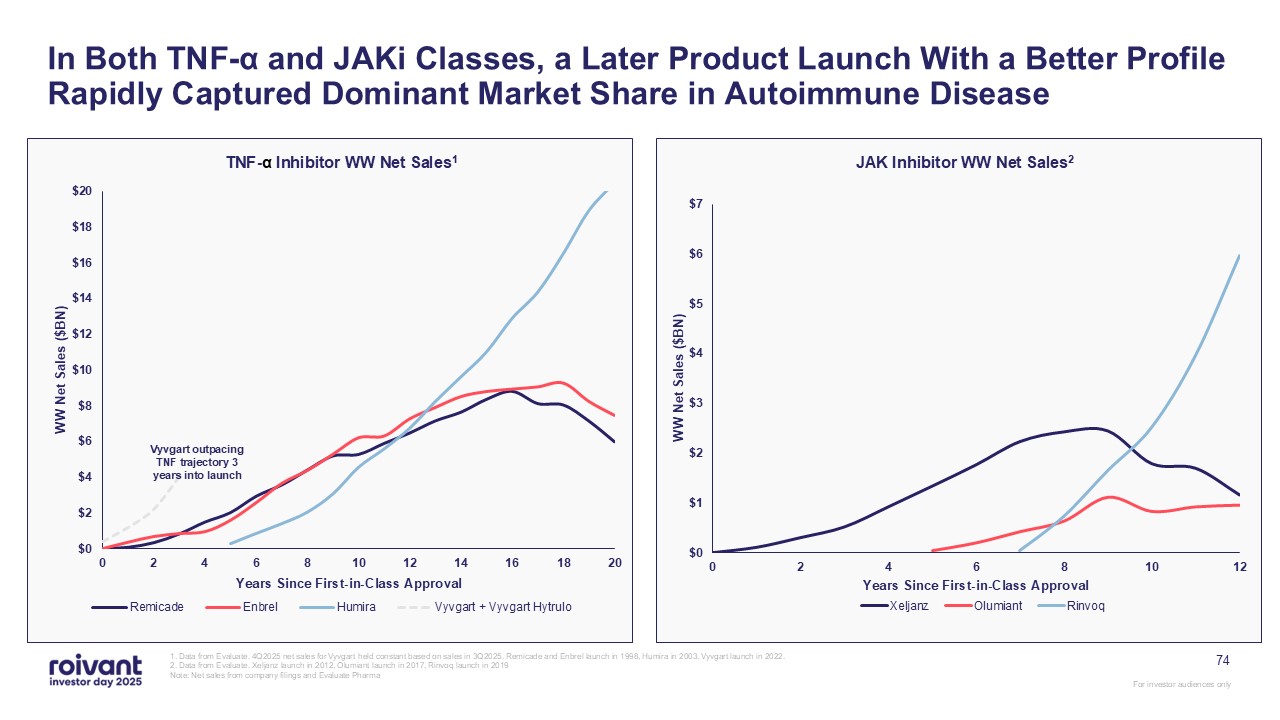
In Both TNF-α and JAKi Classes, a Later Product Launch With a Better Profile
Rapidly Captured Dominant Market Share in Autoimmune Disease 1. Data from Evaluate. 4Q2025 net sales for Vyvgart held constant based on sales in 3Q2025. Remicade and Enbrel launch in 1998, Humira in 2003. Vyvgart launch in 2022. 2. Data
from Evaluate. Xeljanz launch in 2012, Olumiant launch in 2017, Rinvoq launch in 2019 Note: Net sales from company filings and Evaluate Pharma TNF-α Inhibitor WW Net Sales1 JAK Inhibitor WW Net Sales2 74 Vyvgart outpacing TNF trajectory
3 years into launch
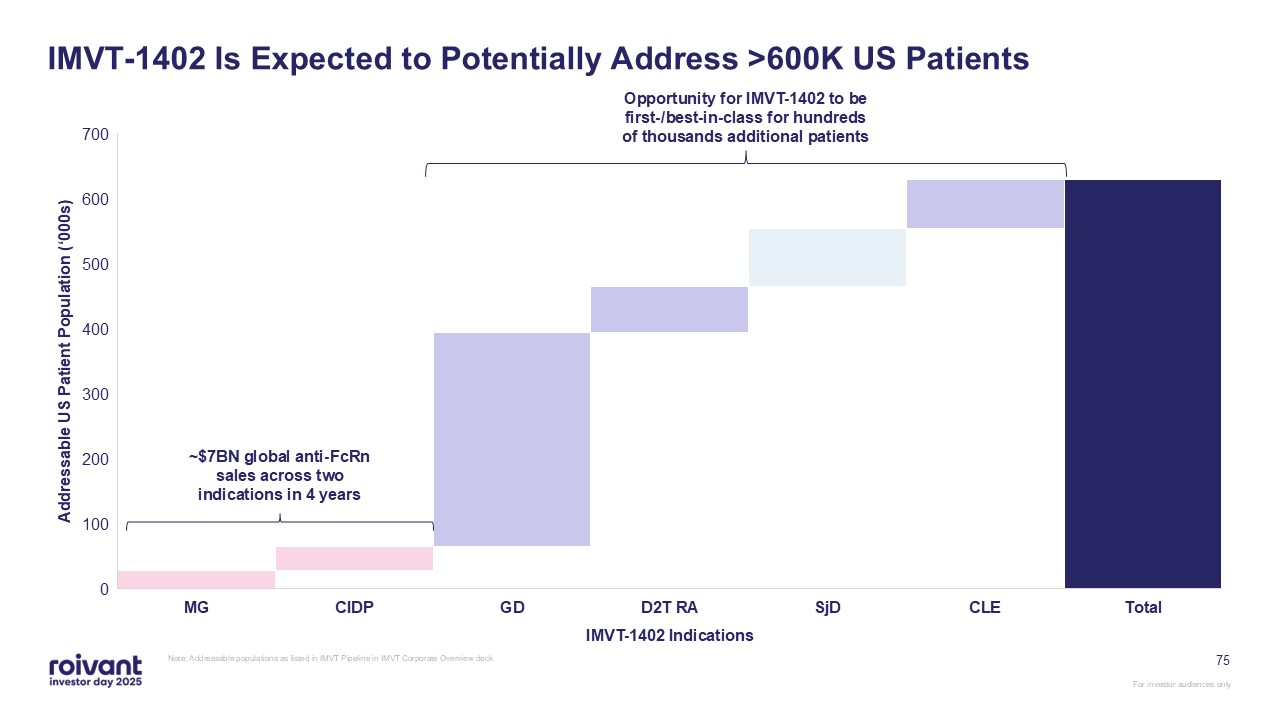
75 IMVT-1402 Is Expected to Potentially Address >600K US Patients Note:
Addressable populations as listed in IMVT Pipeline in IMVT Corporate Overview deck ~$7BN global anti-FcRn sales across two indications in 4 years Opportunity for IMVT-1402 to be first-/best-in-class for hundreds of thousands additional
patients

Near-Term Upside Catalysts for IMVT-1402

77 IMVT-1402 Is Leading in 3 First-/Best-in-Class Indications With Key
Catalysts Expected in D2T RA and CLE in 2026 Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and
may be subject to change. All references are to calendar years. Rapidly enrolling trial; topline results now expected in 2026 (formerly 2027) Difficult-to- Treat Rheumatoid Arthritis Strong PoC data from IMVT-1402 basket study; initial
results from PoC expected in 2026 Cutaneous Lupus Erythematosus Multi-year lead with remarkable PoC data; topline results from both potentially registrational trials expected in 2027 Graves’ Disease
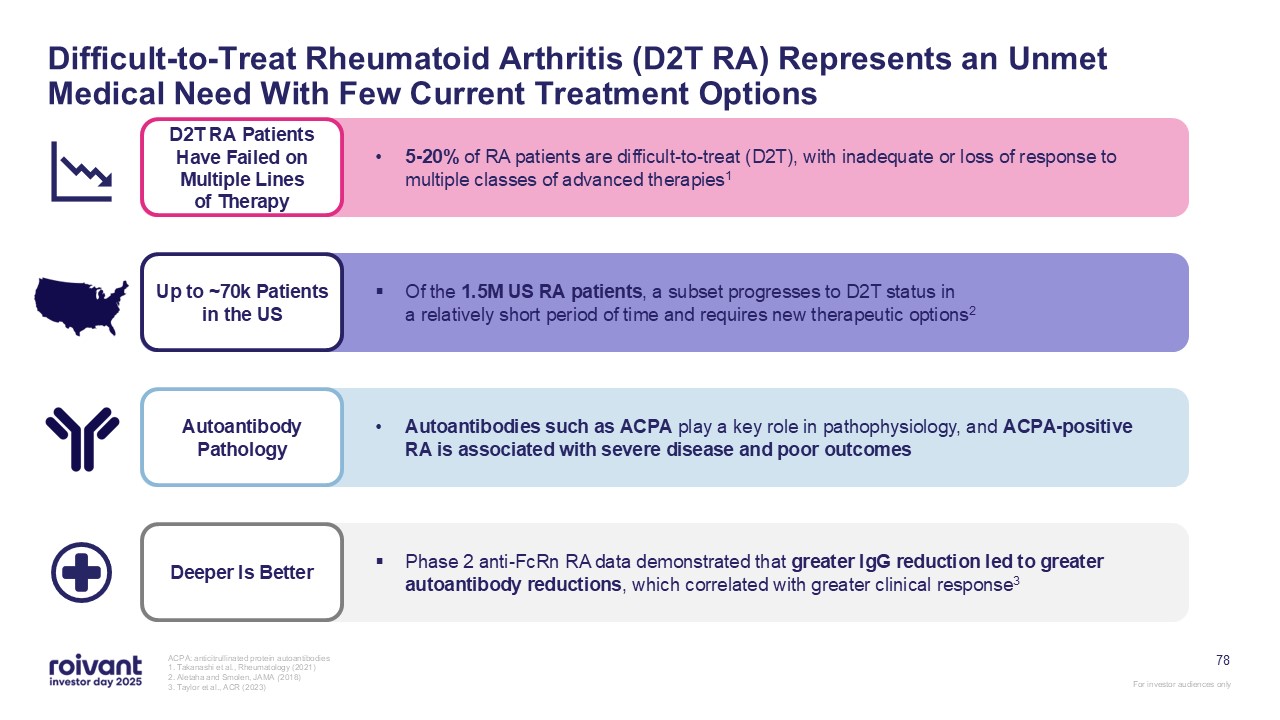
78 Difficult-to-Treat Rheumatoid Arthritis (D2T RA) Represents an Unmet Medical
Need With Few Current Treatment Options ACPA: anticitrullinated protein autoantibodies 1. Takanashi et al., Rheumatology (2021) 2. Aletaha and Smolen, JAMA (2018) 3. Taylor et al., ACR (2023) Deeper Is Better Phase 2 anti-FcRn RA data
demonstrated that greater IgG reduction led to greater autoantibody reductions, which correlated with greater clinical response3 Up to ~70k Patients in the US Of the 1.5M US RA patients, a subset progresses to D2T status ina relatively
short period of time and requires new therapeutic options2 Autoantibody Pathology Autoantibodies such as ACPA play a key role in pathophysiology, and ACPA-positive RA is associated with severe disease and poor outcomes D2T RA Patients
Have Failed on Multiple Lines of Therapy 5-20% of RA patients are difficult-to-treat (D2T), with inadequate or loss of response to multiple classes of advanced therapies1
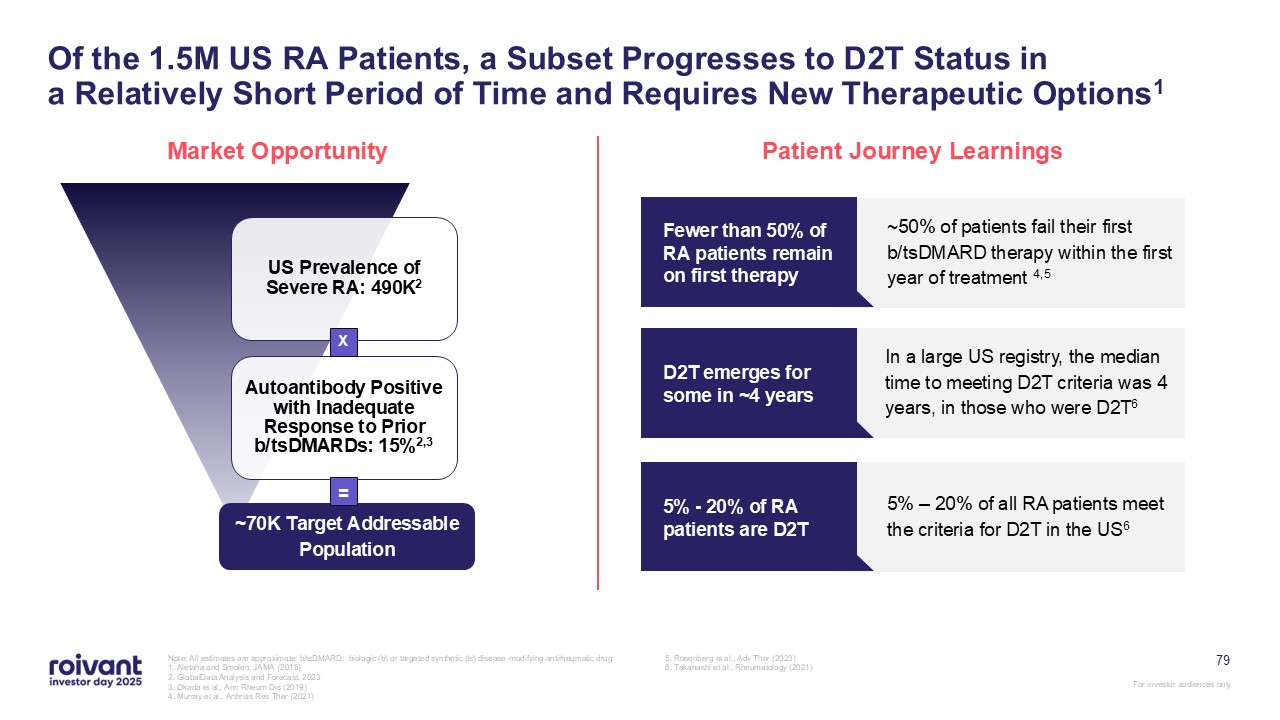
79 Of the 1.5M US RA Patients, a Subset Progresses to D2T Status ina Relatively
Short Period of Time and Requires New Therapeutic Options1 Market Opportunity Patient Journey Learnings ~50% of patients fail their first b/tsDMARD therapy within the first year of treatment 4,5 Fewer than 50% of RA patients remain on
first therapy In a large US registry, the median time to meeting D2T criteria was 4 years, in those who were D2T6 D2T emerges for some in ~4 years 5% – 20% of all RA patients meet the criteria for D2T in the US6 5% - 20% of RA patients
are D2T ~70K Target Addressable Population X = Note: All estimates are approximate; b/tsDMARD: biologic (b) or targeted synthetic (ts) disease-modifying antirheumatic drug 1. Aletaha and Smolen, JAMA (2018) 2. GlobalData Analysis and
Forecast, 2023 3. Okada et al., Ann Rheum Dis (2019) 4. Murray et al., Arthritis Res Ther (2021) 5. Rosenberg et al., Adv Ther (2023) 6. Takanashi et al., Rheumatology (2021)
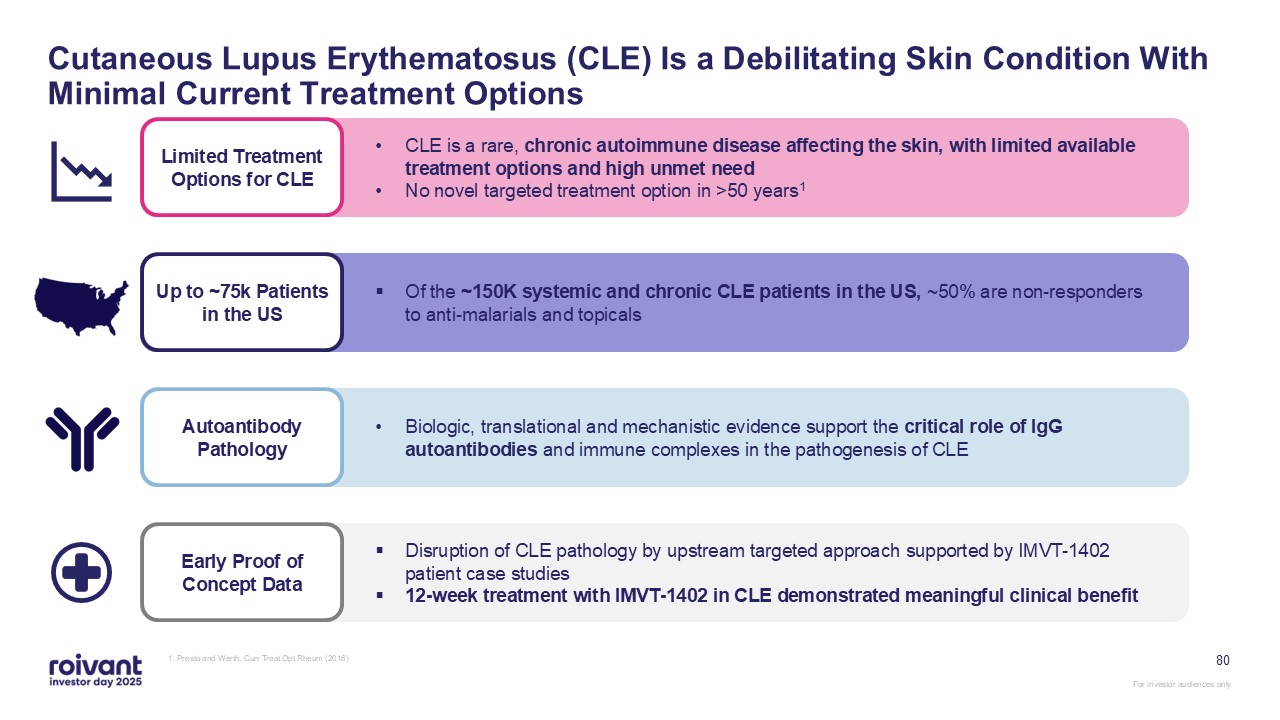
80 Cutaneous Lupus Erythematosus (CLE) Is a Debilitating Skin Condition With
Minimal Current Treatment Options 1. Presto and Werth, Curr Treat Opt Rheum (2016) Early Proof of Concept Data Disruption of CLE pathology by upstream targeted approach supported by IMVT-1402 patient case studies 12-week treatment with
IMVT-1402 in CLE demonstrated meaningful clinical benefit Up to ~75k Patients in the US Of the ~150K systemic and chronic CLE patients in the US, ~50% are non-responders to anti-malarials and topicals Autoantibody Pathology Biologic,
translational and mechanistic evidence support the critical role of IgG autoantibodies and immune complexes in the pathogenesis of CLE Limited Treatment Options for CLE CLE is a rare, chronic autoimmune disease affecting the skin, with
limited available treatment options and high unmet need No novel targeted treatment option in >50 years1
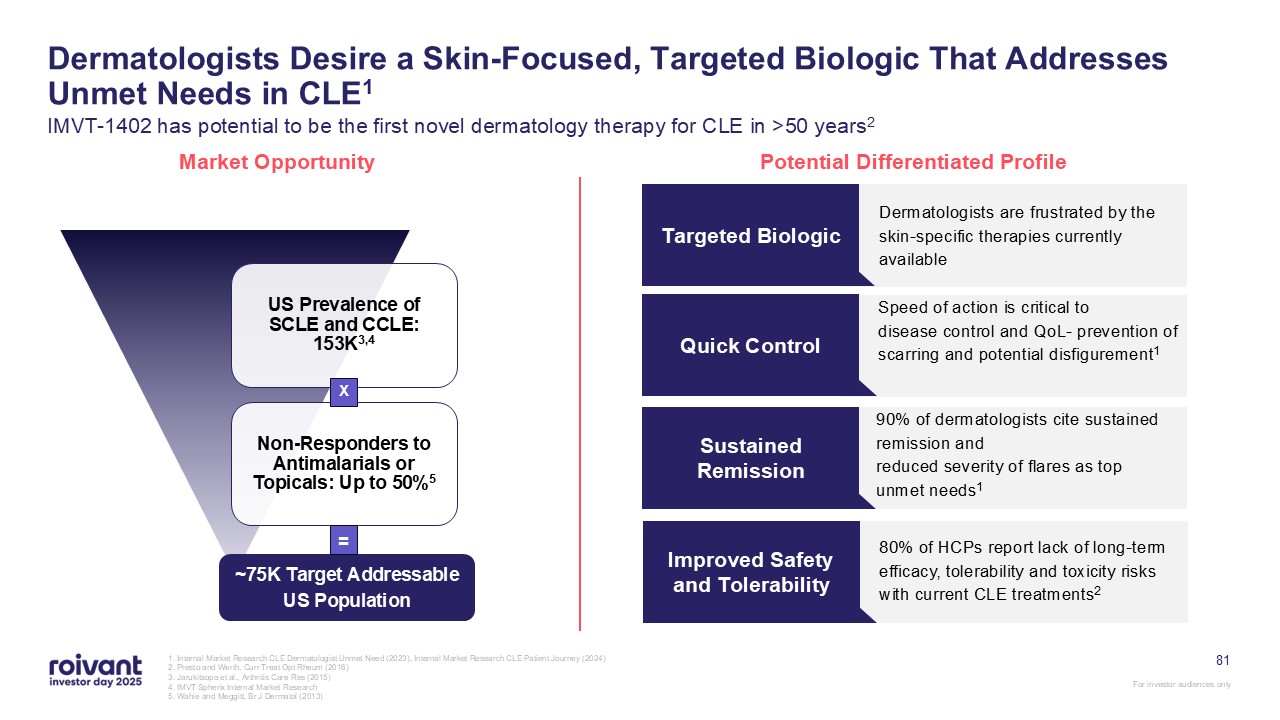
1. Internal Market Research CLE Dermatologist Unmet Need (2023), Internal Market
Research CLE Patient Journey (2024) 2. Presto and Werth, Curr Treat Opt Rheum (2016) 3. Jarukitsopa et al., Arthritis Care Res (2015) 4. IMVT Spherix Internal Market Research 5. Wahie and Meggitt, Br J Dermatol (2013) Dermatologists
Desire a Skin-Focused, Targeted Biologic That Addresses Unmet Needs in CLE1 IMVT-1402 has potential to be the first novel dermatology therapy for CLE in >50 years2 Market Opportunity Potential Differentiated Profile Dermatologists are
frustrated by the skin-specific therapies currently available Targeted Biologic Speed of action is critical to disease control and QoL- prevention of scarring and potential disfigurement1 Quick Control 90% of dermatologists cite sustained
remission and reduced severity of flares as top unmet needs1 Sustained Remission ~75K Target Addressable US Population X = 80% of HCPs report lack of long-term efficacy, tolerability and toxicity risks with current CLE
treatments2 Improved Safety and Tolerability 81

Introducing Dr. Mark LupoGraves’ Disease Thought Leader Mark A. Lupo, MD, FACE,
ECNUThyroid & Endocrine Center of FloridaAssistant Clinical Professor of MedicineFlorida State University, College of MedicineSarasota, Florida

Why Are We Still Treating Graves’ Disease Like It’s 1950? Mark A. Lupo, MD,
FACE, ECNU Thyroid & Endocrine Center of Florida Assistant Clinical Professor of Medicine Florida State University, College of Medicine Sarasota, Florida 83 For investor audiences only

Disclosures Mark A. Lupo, MD Speaking, research, and/or consulting: AbbVie,
Amgen, argenx, Eisai, Immunovant, Interpace Diagnostics, Lycia Therapeutics, QuidelOrtho, Takeda, Viridian 84 For investor audiences only

My Practice Established in 2002 Independent center focused on thyroid and
parathyroid disease 3 Endocrinologists We see/follow hundreds of Graves’ disease patients About half still on long-term antithyroid drug treatment 85 For investor audiences only

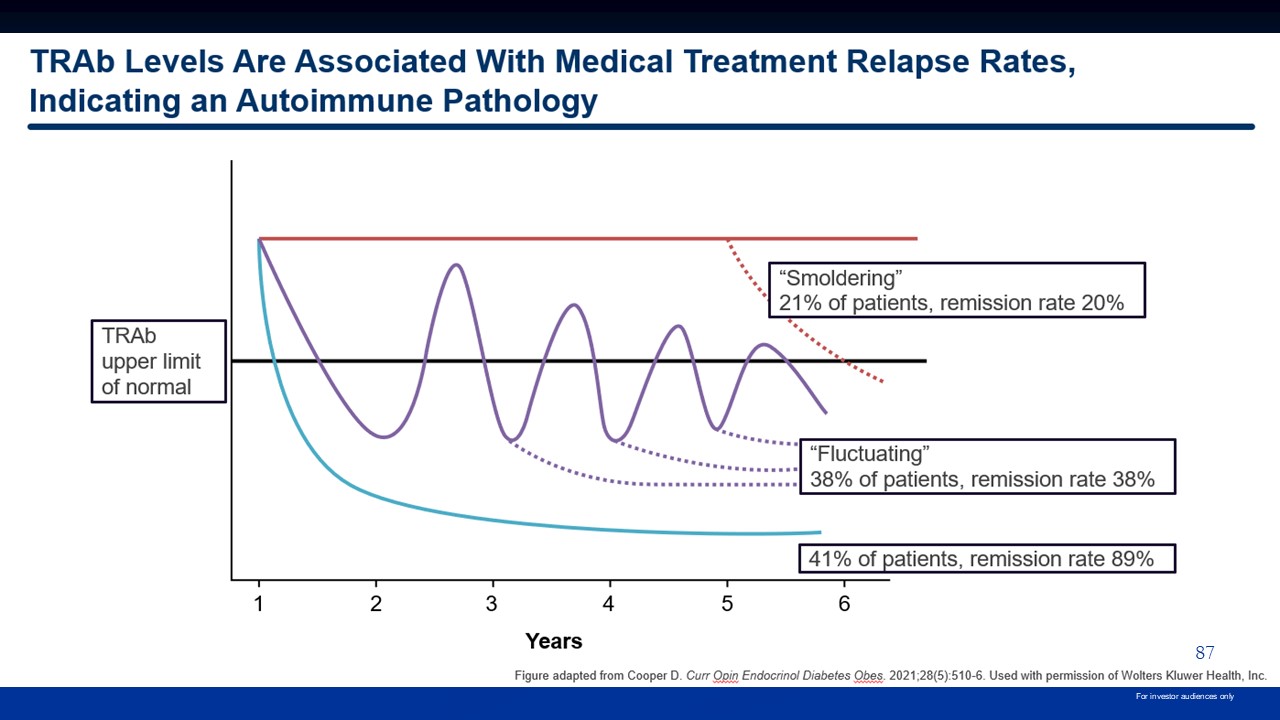
Patient Phenotypes MILD (~50%) Small goiter +/- Slightly high T4/T3 No
TED/mild TED Modest TRAb elevation Predictable ATD response MODERATE (~35-40%) +/- Goiter Overt hyper (T4/T3 elevation) +/- TED mild-moderate TRAb elevation >3-5x normal Multiple ATD dose changes SEVERE (~10-15%) Large
Goiter T4/T3 levels >4-5x normal TED present, often severe TRAb elevation >5x normal High ATD dose with unpredictable responses Factors decreasing remission rates: AGE <40 SEX – male TOBACCO USE 86 For investor audiences
only
87 For investor audiences only

Consequences of Uncontrolled Graves’ Cardiovascular Atrial Fibrillation
Stroke/Death High Output Heart Failure Morbidity/Death Increased Clotting Risks Stroke/Blood Clots Bone Loss Osteoporosis/Fracture Thyroid Eye Disease Vision Threatening Quality of Life Impact Anxiety, Insomnia, Muscle Weakness,
Tremor, Infertility 88 For investor audiences only
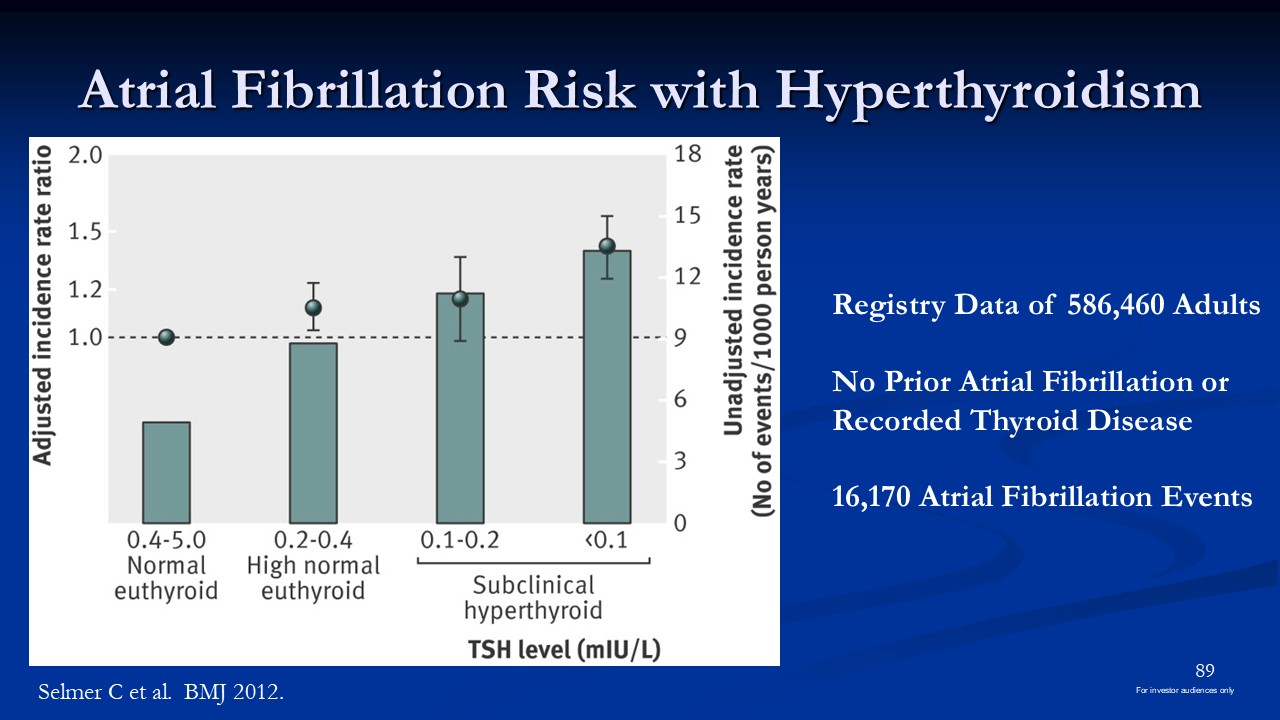
Atrial Fibrillation Risk with Hyperthyroidism Registry Data of 586,460
Adults No Prior Atrial Fibrillation or Recorded Thyroid Disease 16,170 Atrial Fibrillation Events Selmer C et al. BMJ 2012. 89 For investor audiences only
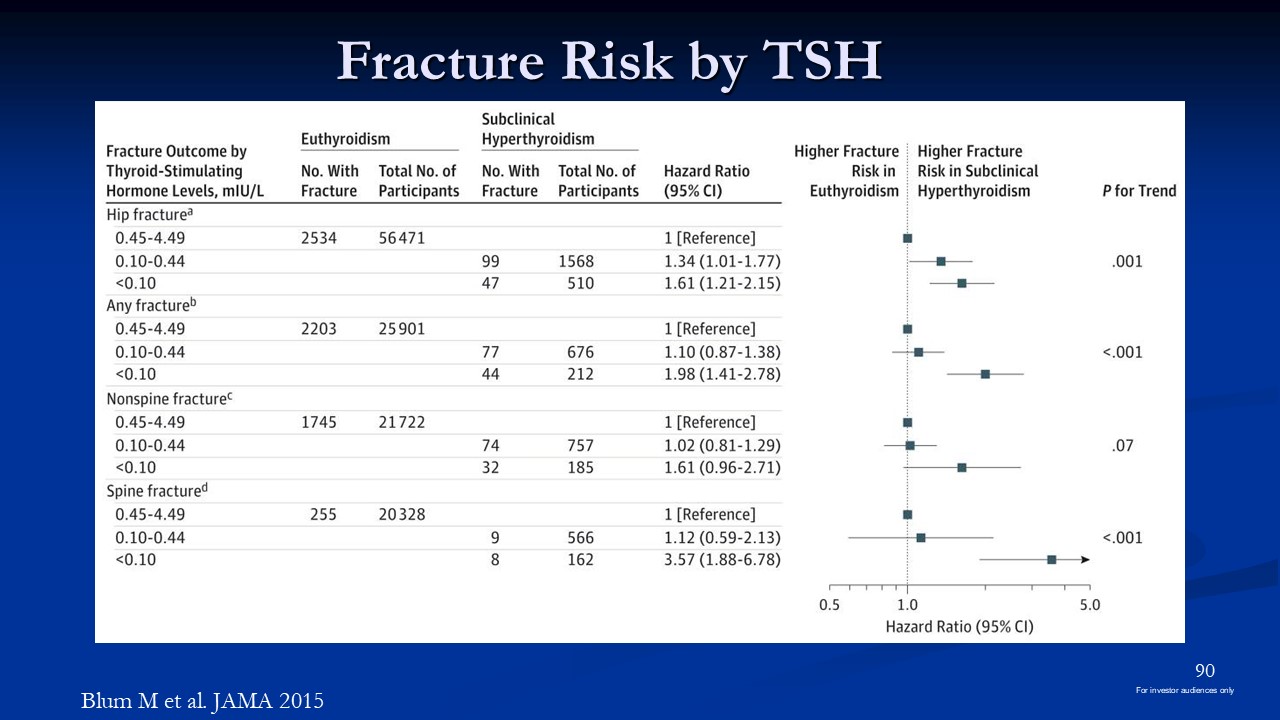
Fracture Risk by TSH Blum M et al. JAMA 2015 90 For investor audiences only
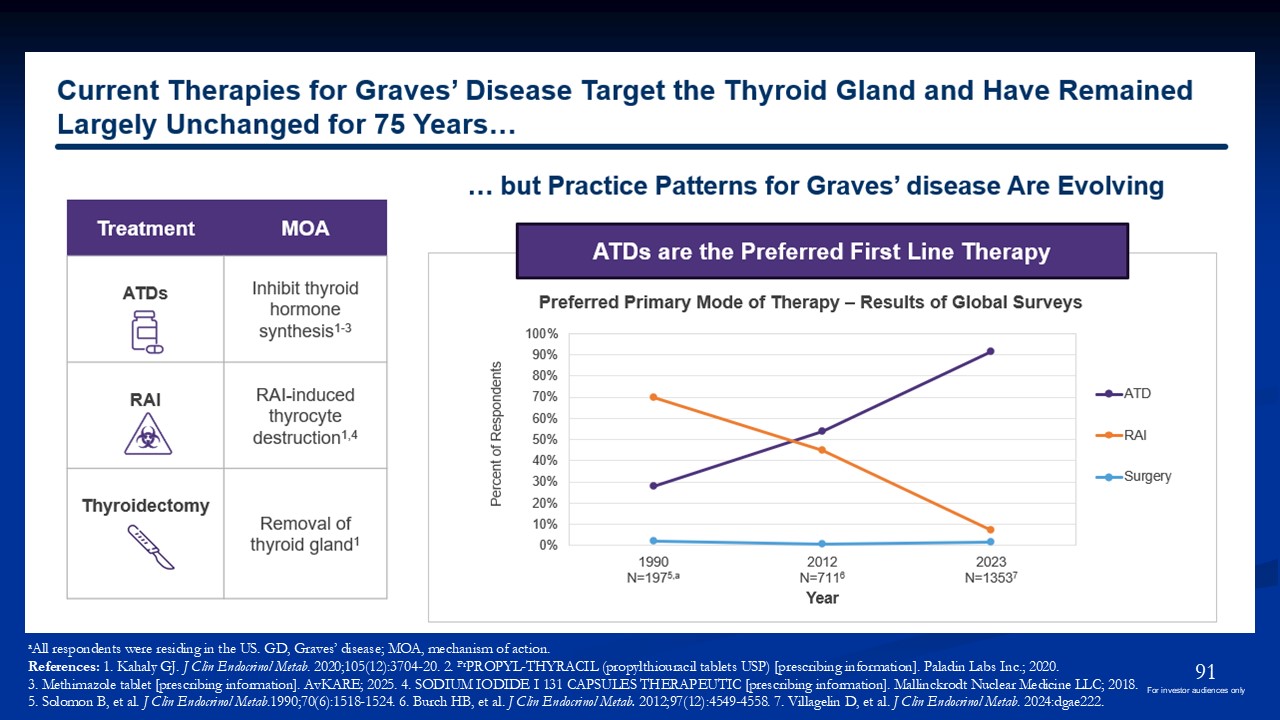
aAll respondents were residing in the US. GD, Graves’ disease; MOA, mechanism of
action. References: 1. Kahaly GJ. J Clin Endocrinol Metab. 2020;105(12):3704-20. 2. PrPROPYL-THYRACIL (propylthiouracil tablets USP) [prescribing information]. Paladin Labs Inc.; 2020. 3. Methimazole tablet [prescribing information].
AvKARE; 2025. 4. SODIUM IODIDE I 131 CAPSULES THERAPEUTIC [prescribing information]. Mallinckrodt Nuclear Medicine LLC; 2018. 5. Solomon B, et al. J Clin Endocrinol Metab.1990;70(6):1518-1524. 6. Burch HB, et al. J Clin Endocrinol Metab.
2012;97(12):4549-4558. 7. Villagelin D, et al. J Clin Endocrinol Metab. 2024:dgae222. 91 For investor audiences only

Definitive Treatment Discussion Radioactive Iodine Increased risk TED TRAb
elevation Radiation exposure Permanent hypothyroidism Thyroidectomy Indicated if concern for cancer or large obstructive goiter Higher risk* Hypoparathyroidism Post-operative bleeding Tracheostomy Scar Permanent
Hypothyroidism *relative to thyroid surgery for other indications 92 For investor audiences only

Quality of Life after Definitive Treatment Hypothyroid patients consistently
report lower scores on QOL scales compared to general population Treatment specific complications 1 in 4 patients still feel “unwell” but often told they are fine due to normal thyroid labs 93 For investor audiences only

Long-term Outcomes 2430 GD patients diagnosed 2003-2005 60% had follow-up data
mean 8 years Remission rates: ATD-45%, RAI-82%, Surgery-96% ATD, second course 29% remission rate Patients receiving ATD had 50% chance of avoiding definitive treatment and 40% chance of achieving euthyroid state Overall, 25% patients
did not feel “fully recovered” long-term Sjolin et al. Thyroid 2019 94 For investor audiences only
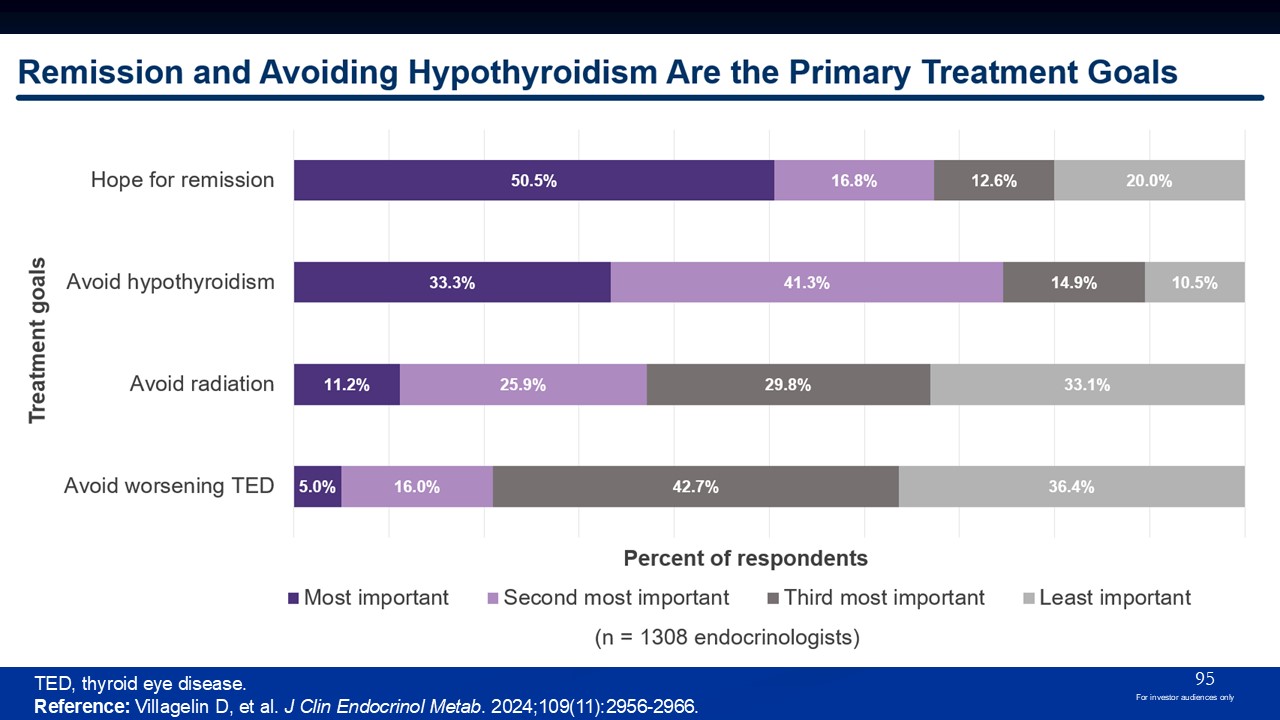
TED, thyroid eye disease. Reference: Villagelin D, et al. J Clin Endocrinol
Metab. 2024;109(11):2956-2966. 95 For investor audiences only
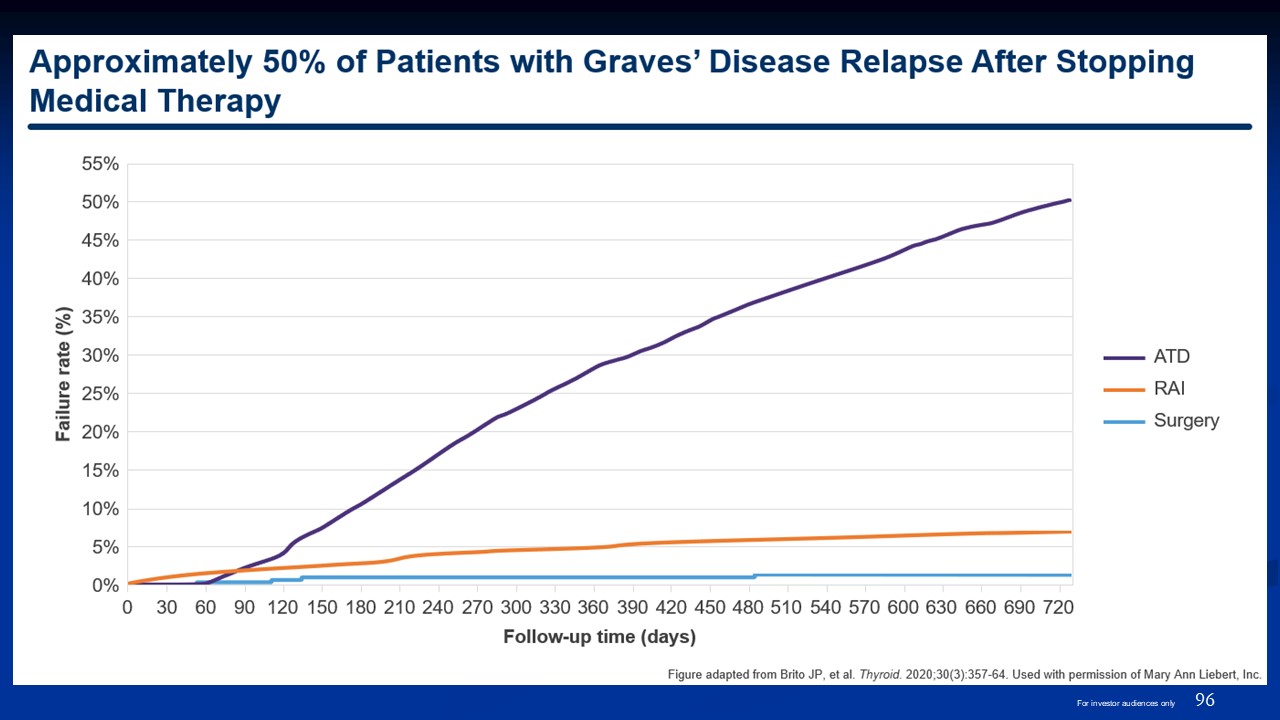
96 For investor audiences only

Real-World Treatment Patterns of Methimazole (MMI) Use in the United
States Study Methods Data Source IQVIA Open Claims and PharMetrics Plus databases Time Period Analyzed November 2017 to October 2023 Inclusion Criteria Patients with a GD diagnosis within 3 years prior to or 2 years after an MMI
prescription Index Date Date of the first MMI prescription claim from November 2020 through October 2021 Follow-Up Assessment Patients were followed for 104 weeks from their first MMI prescription to evaluate treatment patterns Study
Objective To evaluate dosage and treatment patterns following MMI initiation among patients with Graves’ disease in the US GD, Graves’ disease; MMI, methimazole. Lupo m et al. Treatment Patterns Among Methimazole-Treated Patients With
Graves’ Disease. Poster #1112323 Presented at 17th International Thyroid Congress (ITC), June 21, 2025, Rio de Janeiro, Brazil. 97 For investor audiences only
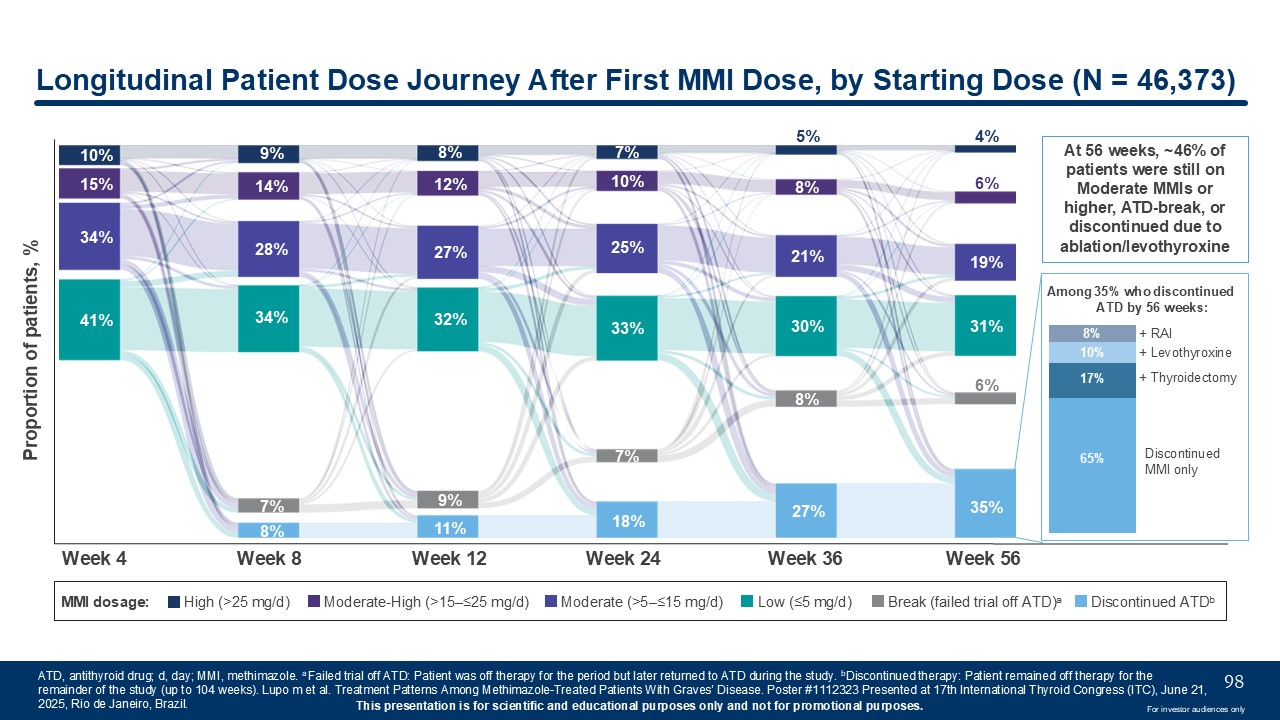
10% 15% 34% 41% 9% 14% 28% 34% 7% 8% 8% 12% 27% 32% 9% 11% 7% 10% 25% 33% 7% 18% 5% 8% 21% 30% 8% 27% 4% 6% 19% 31% 6% 35% Week
4 Week 8 Week 12 Week 24 Week 36 Week 56 Proportion of patients, % MMI dosage: High (>25 mg/d) Moderate-High (>15–≤25 mg/d) Moderate (>5–≤15 mg/d) Low (≤5 mg/d) Break (failed trial off ATD)a Discontinued
ATDb Longitudinal Patient Dose Journey After First MMI Dose, by Starting Dose (N = 46,373) ATD, antithyroid drug; d, day; MMI, methimazole. a Failed trial off ATD: Patient was off therapy for the period but later returned to ATD during the
study. bDiscontinued therapy: Patient remained off therapy for the remainder of the study (up to 104 weeks). Lupo m et al. Treatment Patterns Among Methimazole-Treated Patients With Graves’ Disease. Poster #1112323 Presented at 17th
International Thyroid Congress (ITC), June 21, 2025, Rio de Janeiro, Brazil. 98 Among 35% who discontinued ATD by 56 weeks: 8% 10% 17% 65% Discontinued MMI only + Levothyroxine + RAI + Thyroidectomy At 56 weeks, ~46% of patients
were still on Moderate MMIs or higher, ATD-break, or discontinued due to ablation/levothyroxine For investor audiences only
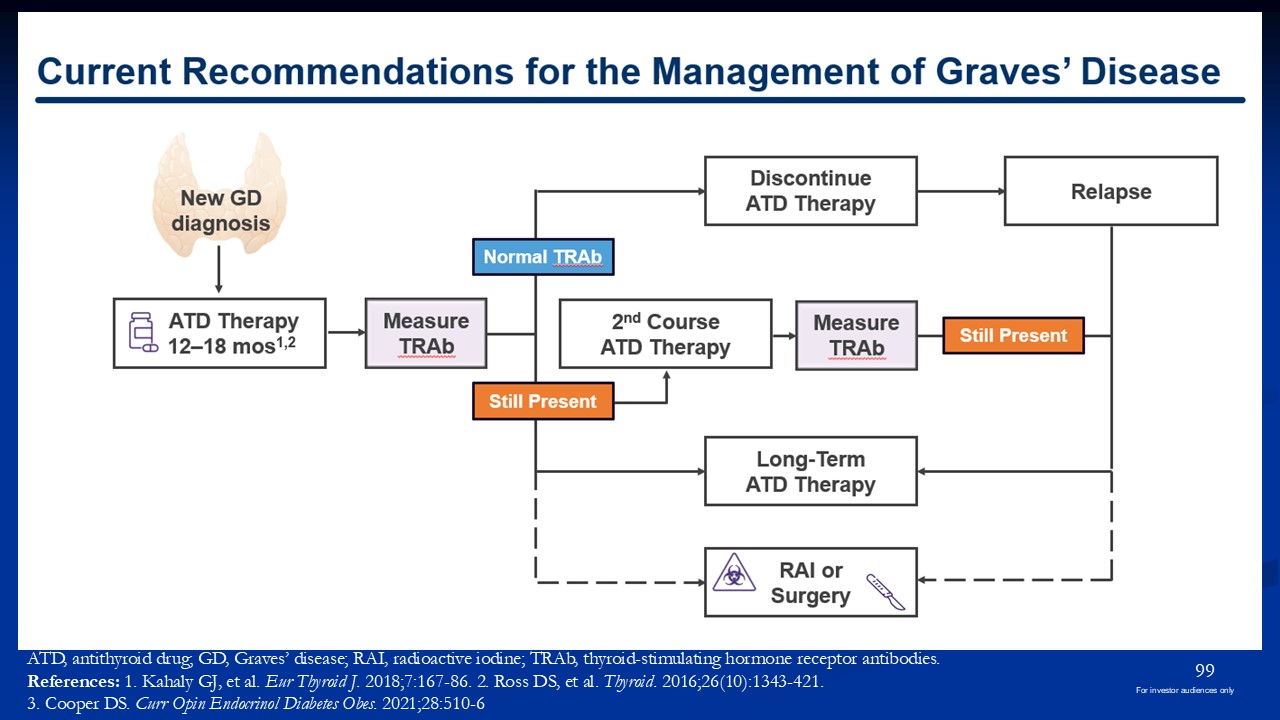
ATD, antithyroid drug; GD, Graves’ disease; RAI, radioactive iodine; TRAb,
thyroid-stimulating hormone receptor antibodies. References: 1. Kahaly GJ, et al. Eur Thyroid J. 2018;7:167-86. 2. Ross DS, et al. Thyroid. 2016;26(10):1343-421. 3. Cooper DS. Curr Opin Endocrinol Diabetes Obes. 2021;28:510-6 99 For
investor audiences only
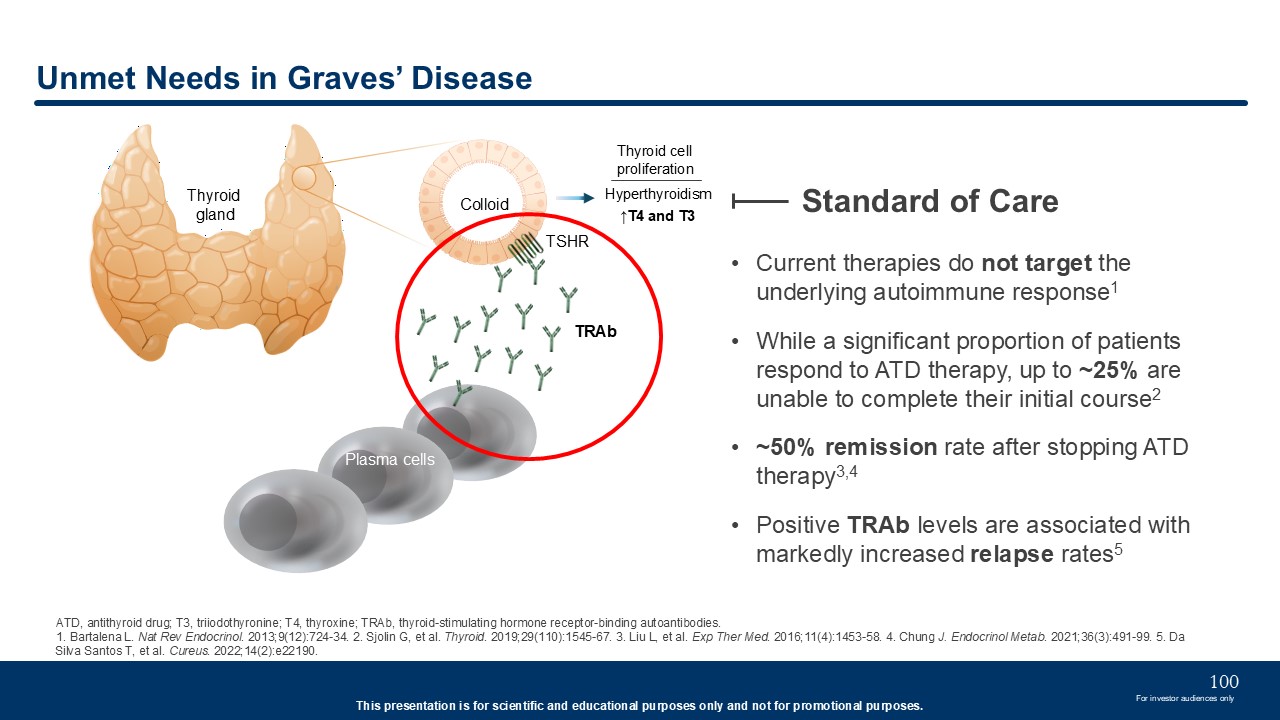
Current therapies do not target the underlying autoimmune response1 While a
significant proportion of patients respond to ATD therapy, up to ~25% are unable to complete their initial course2 ~50% remission rate after stopping ATD therapy3,4 Positive TRAb levels are associated with markedly increased relapse
rates5 Unmet Needs in Graves’ Disease Standard of Care ATD, antithyroid drug; T3, triiodothyronine; T4, thyroxine; TRAb, thyroid-stimulating hormone receptor-binding autoantibodies. 1. Bartalena L. Nat Rev Endocrinol. 2013;9(12):724-34.
2. Sjolin G, et al. Thyroid. 2019;29(110):1545-67. 3. Liu L, et al. Exp Ther Med. 2016;11(4):1453-58. 4. Chung J. Endocrinol Metab. 2021;36(3):491-99. 5. Da Silva Santos T, et al. Cureus. 2022;14(2):e22190. Hyperthyroidism ↑T4 and
T3 Thyroid gland TRAb TSHR Colloid Plasma cells Thyroid cell proliferation 100 For investor audiences only
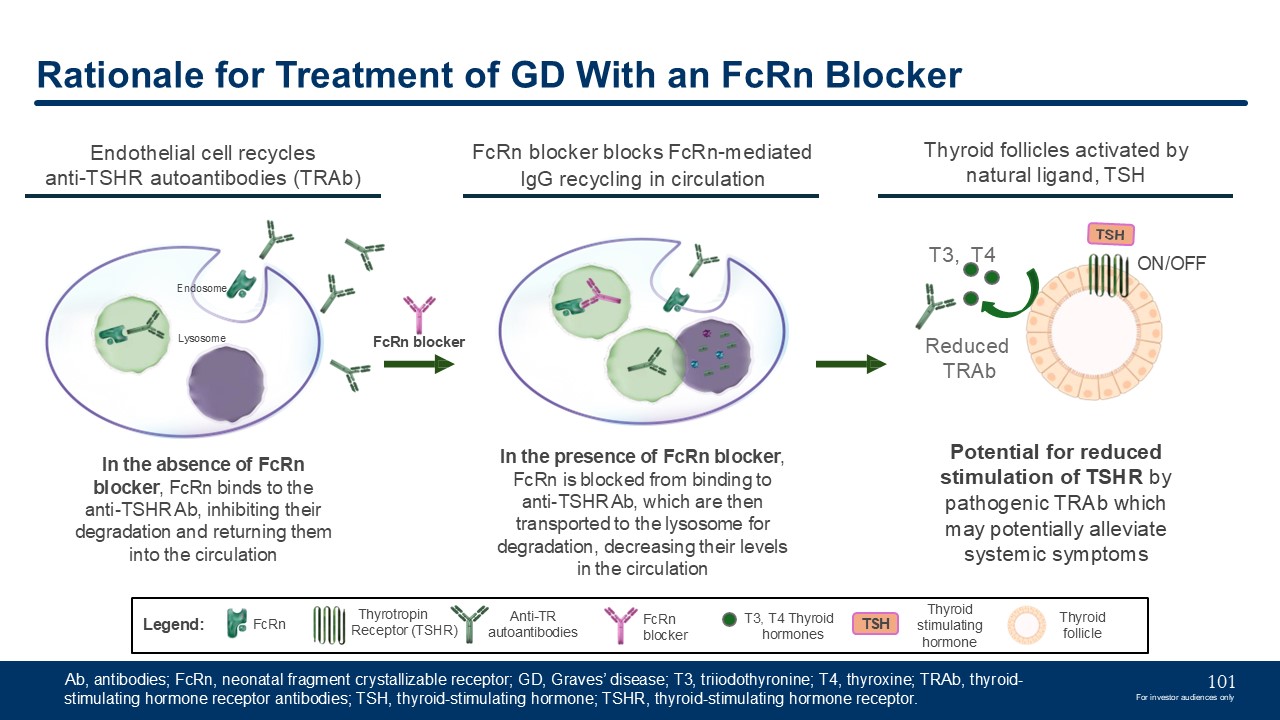
Rationale for Treatment of GD With an FcRn Blocker FcRn blocker blocks
FcRn-mediated IgG recycling in circulation In the presence of FcRn blocker, FcRn is blocked from binding to anti-TSHR Ab, which are then transported to the lysosome for degradation, decreasing their levels in the circulation Lysosome FcRn
blocker Thyroid follicles activated by natural ligand, TSH Reduced TRAb T3, T4 ON/OFF Potential for reduced stimulation of TSHR by pathogenic TRAb which may potentially alleviate systemic symptoms Anti-TR
autoantibodies FcRn Legend: FcRn blocker Thyrotropin Receptor (TSHR) T3, T4 Thyroid hormones TSH Thyroid stimulating hormone In the absence of FcRn blocker, FcRn binds to the anti-TSHR Ab, inhibiting their degradation and returning
them into the circulation Endothelial cell recycles anti-TSHR autoantibodies (TRAb) Thyroid follicle Endosome Ab, antibodies; FcRn, neonatal fragment crystallizable receptor; GD, Graves’ disease; T3, triiodothyronine; T4, thyroxine;
TRAb, thyroid-stimulating hormone receptor antibodies; TSH, thyroid-stimulating hormone; TSHR, thyroid-stimulating hormone receptor. 101 For investor audiences only

Dr. Mark LupoGraves’ Disease Thought Leader Mark A. Lupo, MD, FACE, ECNUThyroid
& Endocrine Center of FloridaAssistant Clinical Professor of MedicineFlorida State University, College of MedicineSarasota, Florida

Paving the Path Forward in Graves’ Disease

Graves’ Disease Patients Have Higher Risk of Sequelae of Severe
Comorbidities 1. Okosieme et al., Lancet Diabetes Endocrinol (2019) 2. Aggarwal et al., Gynecol Obstet Invest (2014) 3. Chin et al., Clin Endocrinol (Oxf.) (2020) 4. Potvin et al., Ophthalmic Plast Reconstr Surg (2023) 5. Galindo et al.,
Thyroid (2019) 6. Bourcier et al., Crit Care Med (2020) 7. Pellegriti et al., J Clin Endocrinol Metab (1998) 7x higher risk1 4x higher risk2 2.5x higher risk1 Relative to Healthy Controls, Graves’ Patients Are at Increased Risk of
Developing Several Severe Comorbidities Untreated or Insufficiently Treated Graves’ Patients Experience Substantial Morbidity and Loss of Quality of Life Thyroid Eye Disease (TED) TED affects ~40% of patients diagnosed with Graves’
disease3 Up to 8% of TED patients experience dysthyroid optic neuropathy (impairment of visual function, leading to permanent sight loss)4 Other Significant Complications In patients hospitalized for Graves’ disease, ~16% are diagnosed
with thyroid storm5, which has a ~20% mortality rate6 Graves’ disease patients who develop thyroid cancer are at a >3x risk of recurrent disease / progressive distant metastases relative to euthyroid controls7 104
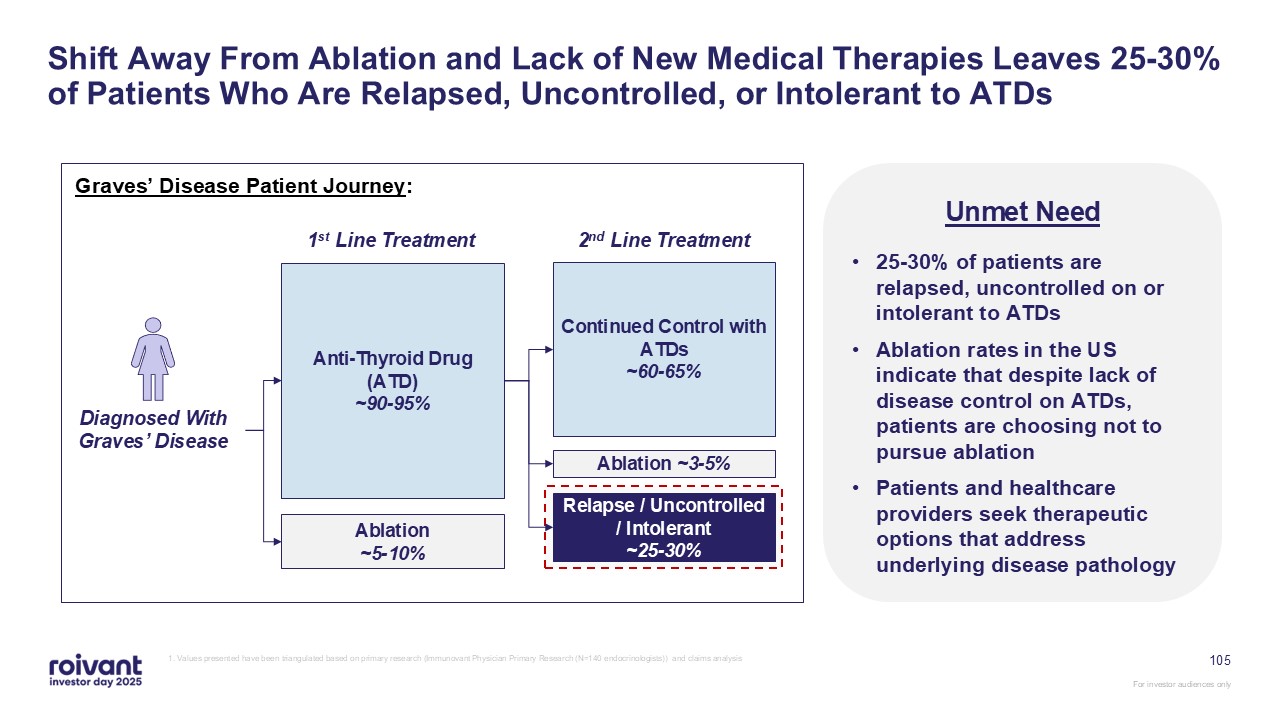
Shift Away From Ablation and Lack of New Medical Therapies Leaves 25-30% of
Patients Who Are Relapsed, Uncontrolled, or Intolerant to ATDs Diagnosed With Graves’ Disease Anti-Thyroid Drug (ATD) ~90-95% Ablation ~5-10% 1st Line Treatment Continued Control with ATDs ~60-65% Ablation ~3-5% Relapse /
Uncontrolled / Intolerant ~25-30% 2nd Line Treatment Graves’ Disease Patient Journey: Unmet Need 25-30% of patients are relapsed, uncontrolled on or intolerant to ATDs Ablation rates in the US indicate that despite lack of disease
control on ATDs, patients are choosing not to pursue ablation Patients and healthcare providers seek therapeutic options that address underlying disease pathology 1. Values presented have been triangulated based on primary research
(Immunovant Physician Primary Research (N=140 endocrinologists)) and claims analysis 105

RAI and surgery are associated with significant complications including
increased risk of death from solid cancers; patients are often hypothyroid and require lifelong thyroid hormone replacement1,2 106 Graves’ Patients Uncontrolled on ATDs Experience Significant Disease Burden and Risk of Adverse Events With
Limited Alternative Treatment Options Chronic ATD use can be associated with risk of severe adverse events, such as hepatotoxicity, pancreatitis, and agranulocytosis (loss of white blood cells)4-6 Uncontrolled Graves’ patients are at risk
for a sequelae of severe comorbidities (e.g., cardiovascular events, thyroid cancer) and experience significant anxiety and impact to quality of life7-8 1. Sundaresh et al., J Clin Endocrinol Metab (2013) 2. Kitahara, et al., JAMA Intern
Med (2019) 3. Suzuki et al., Thyroid (2019) 4. Smith and Hegedüs, N Engl J Med (2016) 5. Brix et al., ETA 2019 6. Okosieme et al., Lancet Diabetes Endocrinol (2019) 7. Cramon et al., Thyroid (2016)
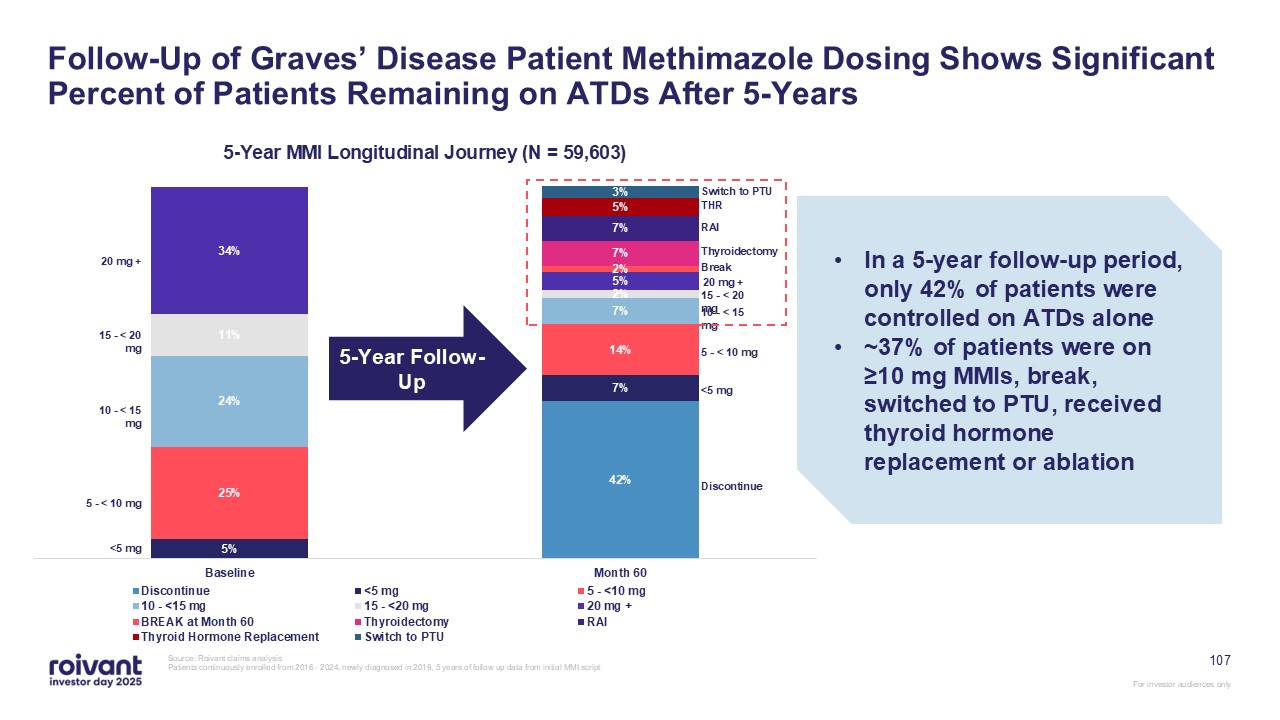
107 Follow-Up of Graves’ Disease Patient Methimazole Dosing Shows Significant
Percent of Patients Remaining on ATDs After 5-Years Source: Roivant claims analysis Patients continuously enrolled from 2016 - 2024, newly diagnosed in 2019, 5 years of follow up data from initial MMI script 5-Year Follow-Up In a 5-year
follow-up period, only 42% of patients were controlled on ATDs alone ~37% of patients were on ≥10 mg MMIs, break, switched to PTU, received thyroid hormone replacement or ablation <5 mg 5 - < 10 mg 10 - < 15 mg 15 - < 20 mg
20 mg + <5 mg 5 - < 10 mg 10 - < 15 mg 15 - < 20 mg 20 mg + Break Switch to PTU THR RAI Thyroidectomy Discontinue
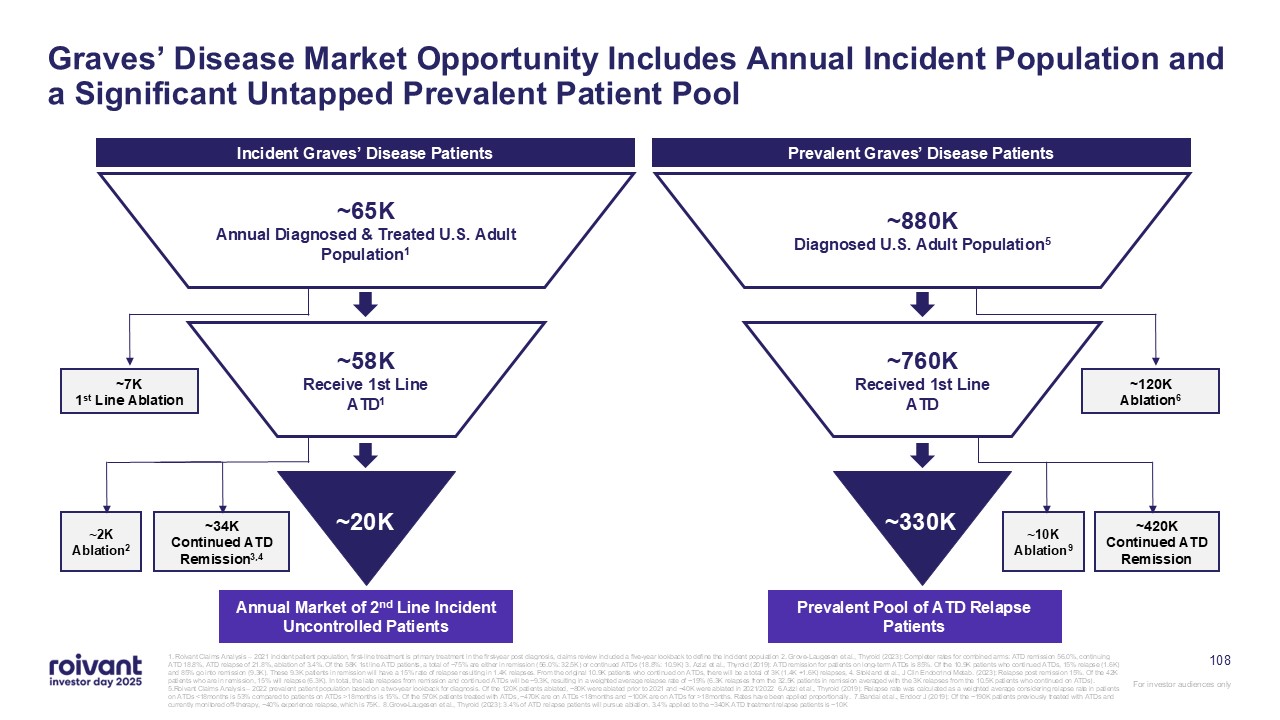
Graves’ Disease Market Opportunity Includes Annual Incident Population and a
Significant Untapped Prevalent Patient Pool Annual Market of 2nd Line Incident Uncontrolled Patients ~7K 1st Line Ablation ~34K Continued ATD Remission3,4 ~65K Annual Diagnosed & Treated U.S. Adult Population1 ~58K Receive 1st
Line ATD1 ~20K ~2K Ablation2 Incident Graves’ Disease Patients Prevalent Pool of ATD Relapse Patients ~120K Ablation6 ~420K Continued ATD Remission ~880K Diagnosed U.S. Adult Population5 ~760K Received 1st Line
ATD ~330K ~10K Ablation9 Prevalent Graves’ Disease Patients 108 1. Roivant Claims Analysis – 2021 incident patient population, first-line treatment is primary treatment in the first-year post diagnosis, claims review included a
five-year lookback to define the incident population 2. Grove-Laugesen et al., Thyroid (2023): Completer rates for combined arms: ATD remission 56.0%, continuing ATD 18.8%, ATD relapse of 21.8%, ablation of 3.4%. Of the 58K 1st line ATD
patients, a total of ~75% are either in remission (56.0%: 32.5K) or continued ATDs (18.8%: 10.9K) 3. Azizi et al., Thyroid (2019): ATD remission for patients on long-term ATDs is 85%. Of the 10.9K patients who continued ATDs, 15% relapse
(1.6K) and 85% go into remission (9.3K). These 9.3K patients in remission will have a 15% rate of relapse resulting in 1.4K relapses. From the original 10.9K patients who continued on ATDs, there will be a total of 3K (1.4K +1.6K) relapses,
4. Stokland et al., J Clin Endocrinol Metab. (2023): Relapse post remission 15%. Of the 42K patients who are in remission, 15% will relapse (6.3K). In total, the late relapses from remission and continued ATDs will be ~9.3K, resulting in a
weighted average relapse rate of ~19% (6.3K relapses from the 32.5K patients in remission averaged with the 3K relapses from the 10.5K patients who continued on ATDs). 5.Roivant Claims Analysis – 2022 prevalent patient population based on a
two-year lookback for diagnosis. Of the 120K patients ablated, ~80K were ablated prior to 2021 and ~40K were ablated in 2021/2022 6.Azizi et al., Thyroid (2019): Relapse rate was calculated as a weighted average considering relapse rate in
patients on ATDs <18months is 53% compared to patients on ATDs >18months is 15%. Of the 570K patients treated with ATDs, ~470K are on ATDs <18months and ~100K are on ATDs for >18months. Rates have been applied proportionally.
7.Bandai et al., Endocr J (2019): Of the ~190K patients previously treated with ATDs and currently monitored off-therapy, ~40% experience relapse, which is 75K. 8.Grove-Laugesen et al., Thyroid (2023): 3.4% of ATD relapse patients will pursue
ablation. 3.4% applied to the ~340K ATD treatment relapse patients is ~10K
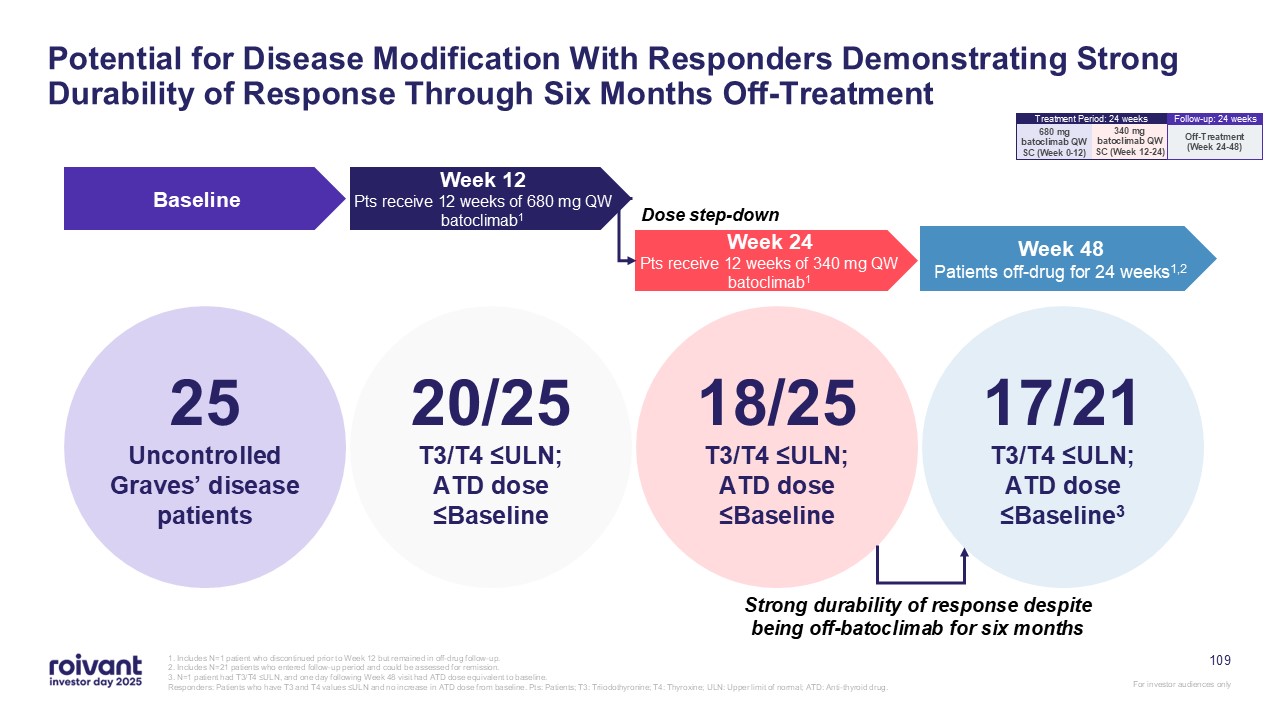
Potential for Disease Modification With Responders Demonstrating Strong
Durability of Response Through Six Months Off-Treatment 25 Uncontrolled Graves’ disease patients Baseline Week 48 Patients off-drug for 24 weeks1,2 Week 12 Pts receive 12 weeks of 680 mg QW batoclimab1 Week 24 Pts receive 12 weeks
of 340 mg QW batoclimab1 20/25 T3/T4 ≤ULN; ATD dose ≤Baseline 18/25 T3/T4 ≤ULN; ATD dose ≤Baseline 17/21 T3/T4 ≤ULN; ATD dose ≤Baseline3 Strong durability of response despite being off-batoclimab for six months Dose step-down 1.
Includes N=1 patient who discontinued prior to Week 12 but remained in off-drug follow-up. 2. Includes N=21 patients who entered follow-up period and could be assessed for remission. 3. N=1 patient had T3/T4 ≤ULN, and one day following
Week 48 visit had ATD dose equivalent to baseline. Responders: Patients who have T3 and T4 values ≤ULN and no increase in ATD dose from baseline. Pts: Patients; T3: Triiodothyronine; T4: Thyroxine; ULN: Upper limit of normal; ATD:
Anti-thyroid drug. 109 340 mg batoclimab QW SC (Week 12-24) 680 mg batoclimab QW SC (Week 0-12) Treatment Period: 24 weeks Off-Treatment (Week 24-48) Follow-up: 24 weeks
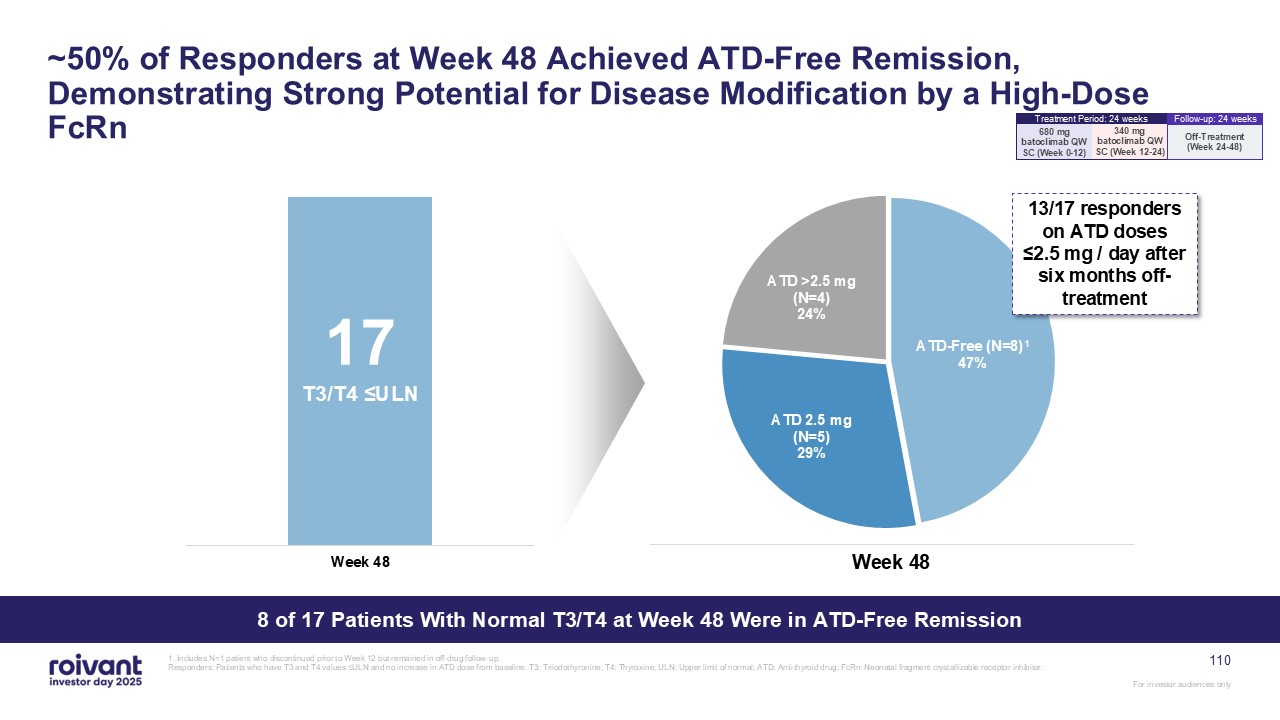
~50% of Responders at Week 48 Achieved ATD-Free Remission, Demonstrating Strong
Potential for Disease Modification by a High-Dose FcRn 8 of 17 Patients With Normal T3/T4 at Week 48 Were in ATD-Free Remission 17 T3/T4 ≤ULN Week 48 13/17 responders on ATD doses ≤2.5 mg / day after six months off-treatment 1.
Includes N=1 patient who discontinued prior to Week 12 but remained in off-drug follow-up. Responders: Patients who have T3 and T4 values ≤ULN and no increase in ATD dose from baseline. T3: Triiodothyronine; T4: Thyroxine; ULN: Upper limit
of normal; ATD: Anti-thyroid drug; FcRn: Neonatal fragment crystallizable receptor inhibitor. 110 340 mg batoclimab QW SC (Week 12-24) 680 mg batoclimab QW SC (Week 0-12) Treatment Period: 24 weeks Off-Treatment (Week
24-48) Follow-up: 24 weeks
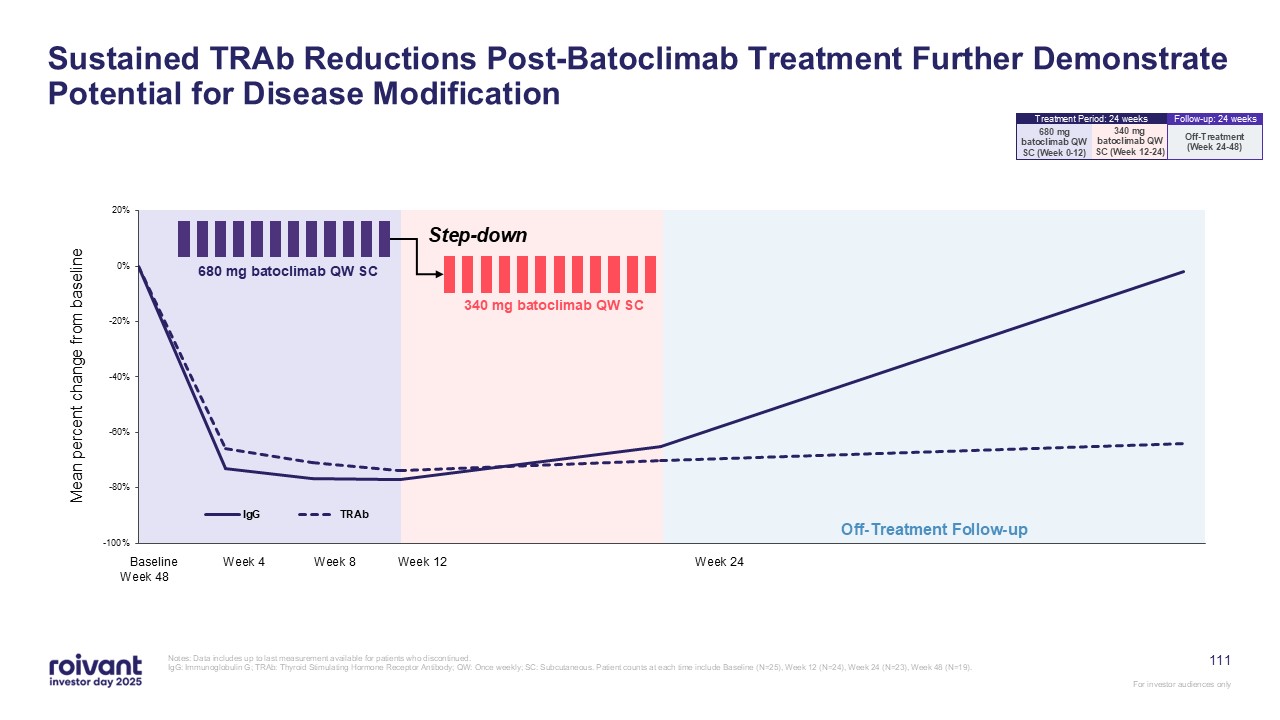
Off-Treatment Follow-up Mean percent change from baseline Baseline Week 4
Week 8 Week 12 Week 24 Week 48 340 mg batoclimab QW SC 680 mg batoclimab QW SC Step-down Sustained TRAb Reductions Post-Batoclimab Treatment Further Demonstrate Potential for Disease Modification Notes: Data includes up to last
measurement available for patients who discontinued. IgG: Immunoglobulin G; TRAb: Thyroid Stimulating Hormone Receptor Antibody; QW: Once weekly; SC: Subcutaneous. Patient counts at each time include Baseline (N=25), Week 12 (N=24), Week 24
(N=23), Week 48 (N=19). 111 340 mg batoclimab QW SC (Week 12-24) 680 mg batoclimab QW SC (Week 0-12) Treatment Period: 24 weeks Off-Treatment (Week 24-48) Follow-up: 24 weeks

112 IMVT-1402 Could Potentially Be the First-in-Class Disease-Modifying Therapy
in Graves’ Disease Remarkable effect seen in uncontrolled Graves’ disease patients: 18 of 25 patients treated with batoclimab are responders at Week 24 01 Durable off-drug response: of the 21 patients who entered the off-drug follow-up
period, 17 remain responders six months following batoclimab treatment 02 IMVT-1402 pivotal trial design could potentially generate improved efficacy data due to continuous 600 mg QW dosing vs. batoclimab’s step-down dosing design 04 Two
potentially registrational trials for IMVT-1402 in Graves’ disease currently enrolling 05 First-ever observed ATD-free remission in uncontrolled patients: 8 of 17 responders remain off all medications six months following batoclimab
treatment demonstrating potential for disease modification 03 Notes: Data includes N=1 patient who discontinued prior to Week 12 but remained in off-drug follow-up. ATD: Anti-thyroid drug; QW: Once weekly; responders: patients who have T3
and T4 values ≤ULN and no increase in ATD dose from baseline.

Opportunities for IMVT-1402 to Win on Efficacy

114 IMVT-1402 Has the Potential to Be Best-in-Class in MG, SjD and CIDP, With
Room to Penetrate Large, Well-Validated Markets Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback,
and may be subject to change. All references are to calendar years. Market rapidly expanding with space for multiple blockbuster agents; topline results expected in 2027 Myasthenia Gravis Expected to be best-in-class with limited
entrenched competition; topline results expected in 2028 Sjögren’s Disease Market quickly growing with 1 approved agent; topline results expected in 2028 Chronic Inflammatory Demyelinating Polyneuropathy
115 Sjögren’s Disease (SjD) Is a Chronic Autoimmune Disease Characterized by
Lymphocytic Infiltration of the Salivary and Lacrimal Glands 1. Mariette et al., N. Engl J Med (2018) 2. Brito-Zeron et al., Nat Rev Dis Primers (2016) 3. GlobalData Analysis and Forecast, January 2025 4. Gottenberg et al., ACR
(2024) Deeper Is Better Nipocalimab data demonstrated that deeper IgG reduction leads to better clinical response across all primary and secondary endpoints4 Up to ~90k Addressable Patients in the US Of the ~290K primary SjD patients in
the US, ~30% are moderate-severe with anti-Ro/SSA antibodies3 Autoantibody Pathology Autoantibodies detected in ~50-70% of patients with primary SjD; anti-FcRn proof of mechanism established Limited Treatment Options for SjD SjD symptoms
include severe dryness of the eyes and mouth; the latter frequently associated with difficulty swallowing or speaking, tooth decay, gum disease, and impaired QoL1,2 No therapies approved for the treatment of primary Sjogren’s disease
Sizable SjD Patient Group With Unmet Need for an Approved Treatment
Option Sizable Unmet Need Expansion Opportunities Potential to impact conditions with shared autoimmune pathology Secondary Sjögren’s Unmet need to improve glandular manifestations beyond symptom relief Glandular Disease Disease impact
on patient QoL varies widely; so-called “nuisance” symptoms can become debilitating if inadequately managed Less Severe Disease ~90K Target Addressable US Population X = 116 Note: All estimates are approximate 1. GlobalData Analysis
and Forecast, January 2025 2. Brito-Zeron P et al., Nat Rev Dis Primers (2016) 3. Decision Resources Group
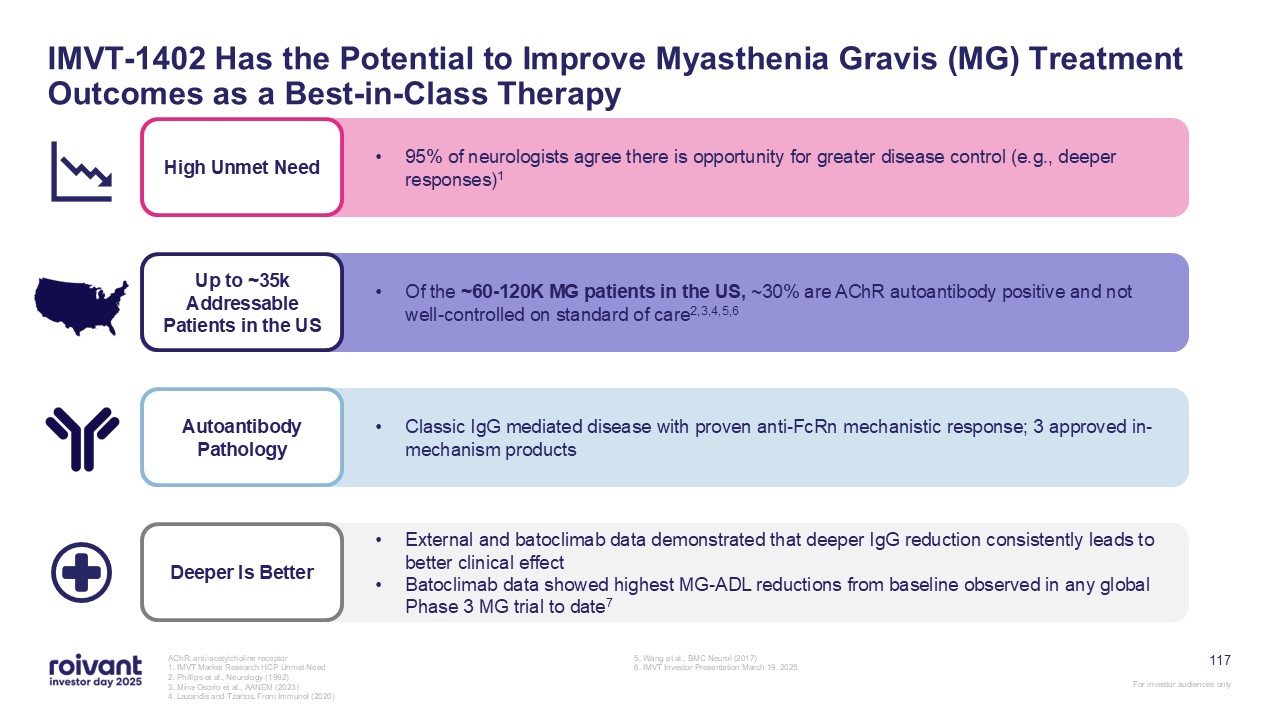
117 IMVT-1402 Has the Potential to Improve Myasthenia Gravis (MG) Treatment
Outcomes as a Best-in-Class Therapy AChR: anti-acetylcholine receptor 1. IMVT Market Research HCP Unmet Need 2. Phillips et al., Neurology (1992) 3. Mina-Osorio et al., AANEM (2023) 4. Lazaridis and Tzartos, Front Immunol (2020) 5.
Wang et al., BMC Neurol (2017) 6. IMVT Investor Presentation March 19, 2025 Deeper Is Better External and batoclimab data demonstrated that deeper IgG reduction consistently leads to better clinical effect Batoclimab data showed highest
MG-ADL reductions from baseline observed in any global Phase 3 MG trial to date7 Up to ~35k Addressable Patients in the US Of the ~60-120K MG patients in the US, ~30% are AChR autoantibody positive and not well-controlled on standard of
care2,3,4,5,6 Autoantibody Pathology Classic IgG mediated disease with proven anti-FcRn mechanistic response; 3 approved in-mechanism products High Unmet Need 95% of neurologists agree there is opportunity for greater disease control
(e.g., deeper responses)1
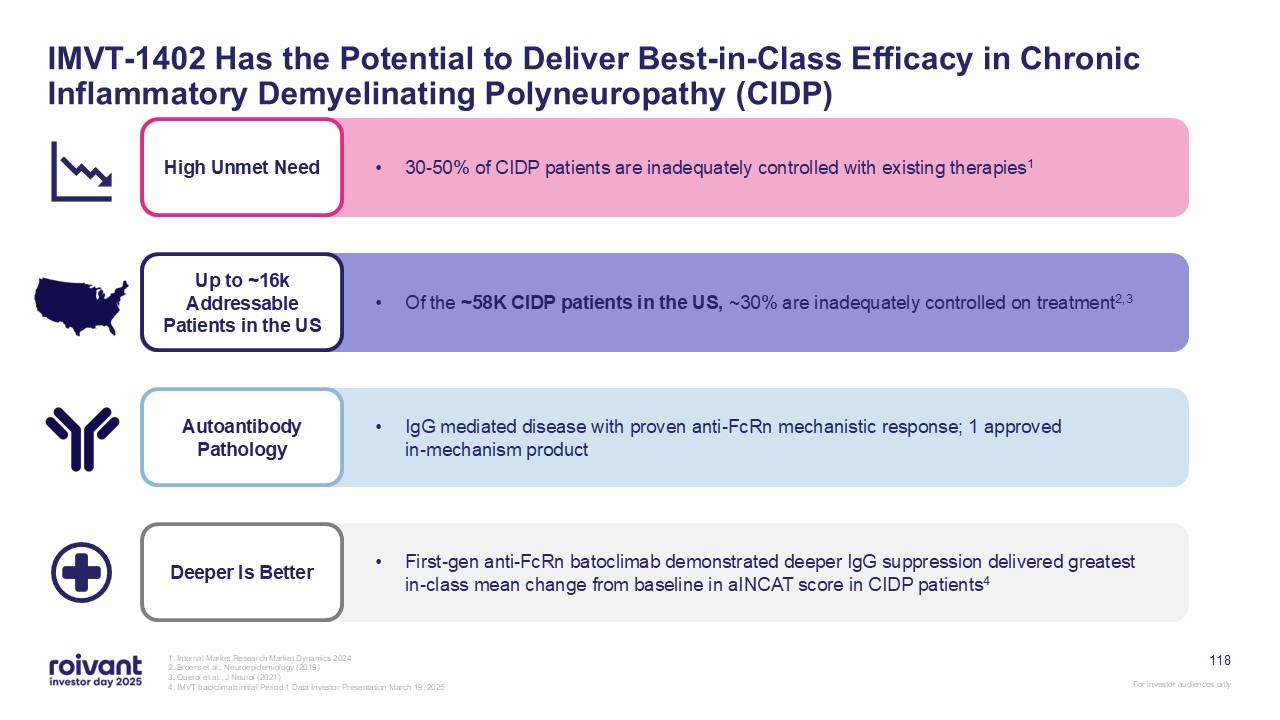
118 IMVT-1402 Has the Potential to Deliver Best-in-Class Efficacy in Chronic
Inflammatory Demyelinating Polyneuropathy (CIDP) 1. Internal Market Research Market Dynamics 2024 2. Broers et al., Neuroepidemiology (2019) 3. Querol et al., J Neurol (2021) 4. IMVT batoclimab initial Period 1 Data Investor Presentation
March 19, 2025 Deeper Is Better First-gen anti-FcRn batoclimab demonstrated deeper IgG suppression delivered greatest in-class mean change from baseline in aINCAT score in CIDP patients4 Up to ~16k Addressable Patients in the US Of the
~58K CIDP patients in the US, ~30% are inadequately controlled on treatment2,3 Autoantibody Pathology IgG mediated disease with proven anti-FcRn mechanistic response; 1 approved in-mechanism product High Unmet Need 30-50% of CIDP patients
are inadequately controlled with existing therapies1
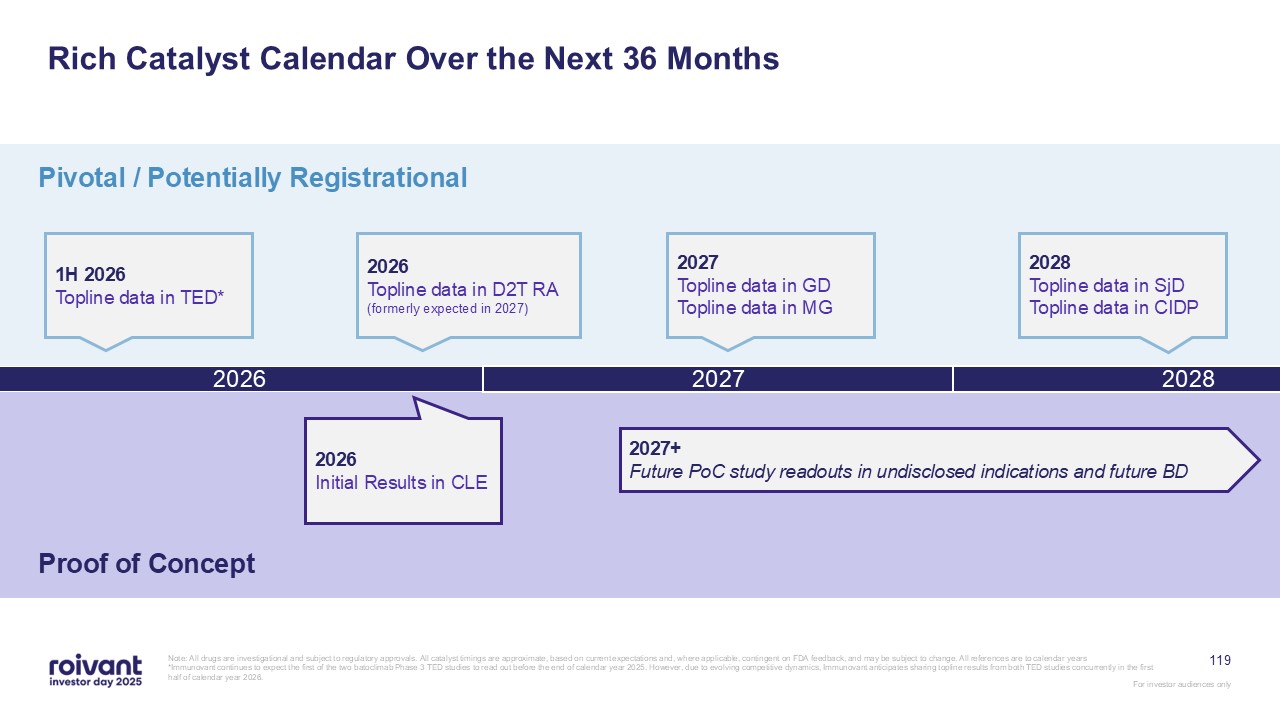
Rich Catalyst Calendar Over the Next 36 Months 2026 Pivotal / Potentially
Registrational Proof of Concept 1H 2026 Topline data in TED* Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on
FDA feedback, and may be subject to change. All references are to calendar years *Immunovant continues to expect the first of the two batoclimab Phase 3 TED studies to read out before the end of calendar year 2025. However, due to evolving
competitive dynamics, Immunovant anticipates sharing topline results from both TED studies concurrently in the first half of calendar year 2026. 2027 2028 2026 Topline data in D2T RA (formerly expected in 2027) 2026 Initial Results in
CLE 2027 Topline data in GD Topline data in MG 2028 Topline data in SjD Topline data in CIDP 119 2027+ Future PoC study readouts in undisclosed indications and future BD

120 In Summary: IMVT-1402 Focused clinical execution: topline data in D2T RA
now expected in 2026; readouts in 3 potentially registrational trials and 1 PoC expected in next 24 months The anti-FcRn class is rapidly growing; precedent best-in-class products have won significant market share in I&I
indications Graves’ disease has extraordinary unmet need; we have demonstrated best-in-class potential with a multi-year lead Multiple shots on goal: IMVT-1402 offers best-in-class profile in 5 late-stage indications and 1 PoC Note: All
drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar
years. For investor audiences only
Q&A

122 Mosliciguat Drew Fromkin CEO, Pulmovant Frank Torti President &
Vant Chair, Roivant

123 PH-ILD represents an area of intense unmet medical need with only one
approved mechanism (two therapies) and an estimated 200,000 patients across the US and Europe Topline data from ongoing Phase 2 study (PHocus) is expected in 2H 2026 – 120 patient study with the potential to define a new standard of care in
PH-ILD Mosliciguat with a differentiated mechanism of action – inhaled soluble guanylate cyclase (sGC) activator – is potentially the first non-treprostinil treatment option for PH-ILD patients Parallels to PAH market with combination
therapies present across the disease spectrum; however, PH-ILD expected to be larger commercial opportunity with competition limited to inhaled mechanisms Among the best PVR reductions seen to date with convenient once-daily dosing and
favorable safety profile across 170 healthy volunteers and PH patients – approved drugs have shown PVR reductions translate to clinical efficacy Key Takeaways: Mosliciguat For investor audiences only PH-ILD: Pulmonary hypertension
associated with interstitial lung disease Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may
be subject to change. All references are to calendar years.
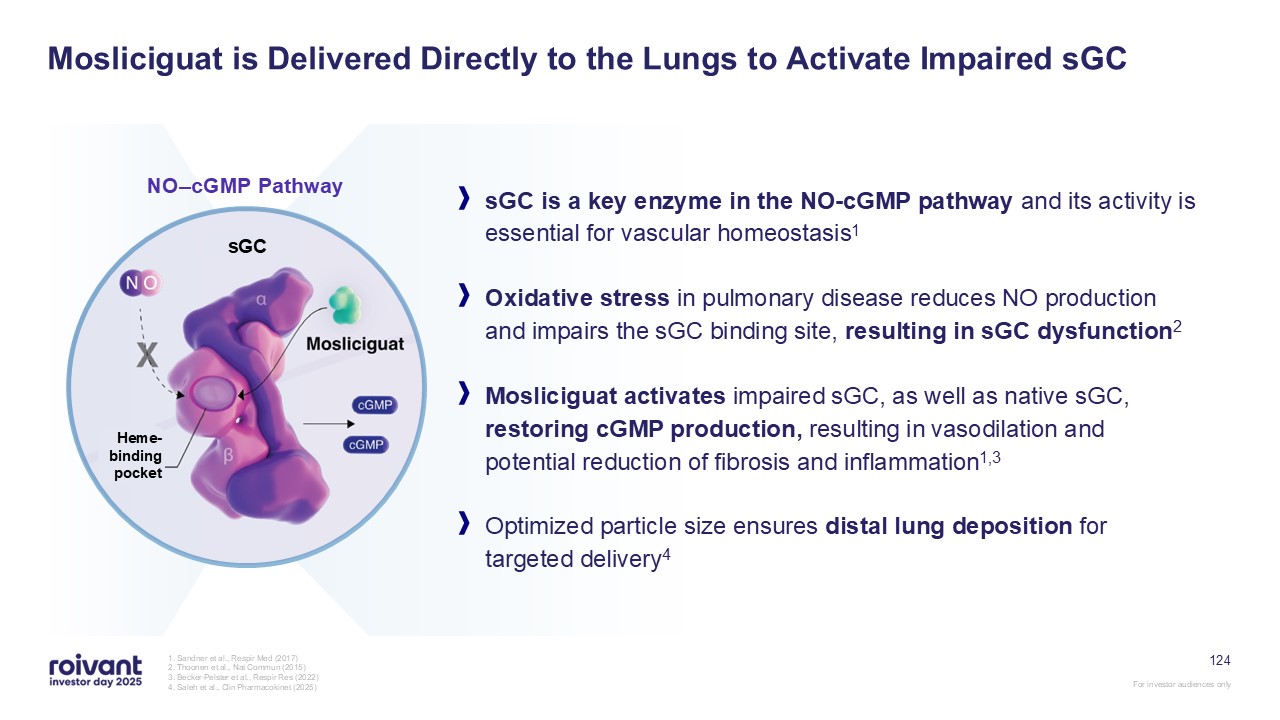
124 1. Sandner et al., Respir Med (2017) 2. Thoonen et al., Nat Commun
(2015) 3. Becker-Pelster et al., Respir Res (2022) 4. Saleh et al., Clin Pharmacokinet (2025) x x sGC is a key enzyme in the NO-cGMP pathway and its activity is essential for vascular homeostasis1 Oxidative stress in pulmonary disease
reduces NO production and impairs the sGC binding site, resulting in sGC dysfunction2 Mosliciguat activates impaired sGC, as well as native sGC, restoring cGMP production, resulting in vasodilation and potential reduction of fibrosis and
inflammation1,3 Optimized particle size ensures distal lung deposition for targeted delivery4 Mosliciguat is Delivered Directly to the Lungs to Activate Impaired sGC NO–cGMP Pathway Heme-binding pocket sGC
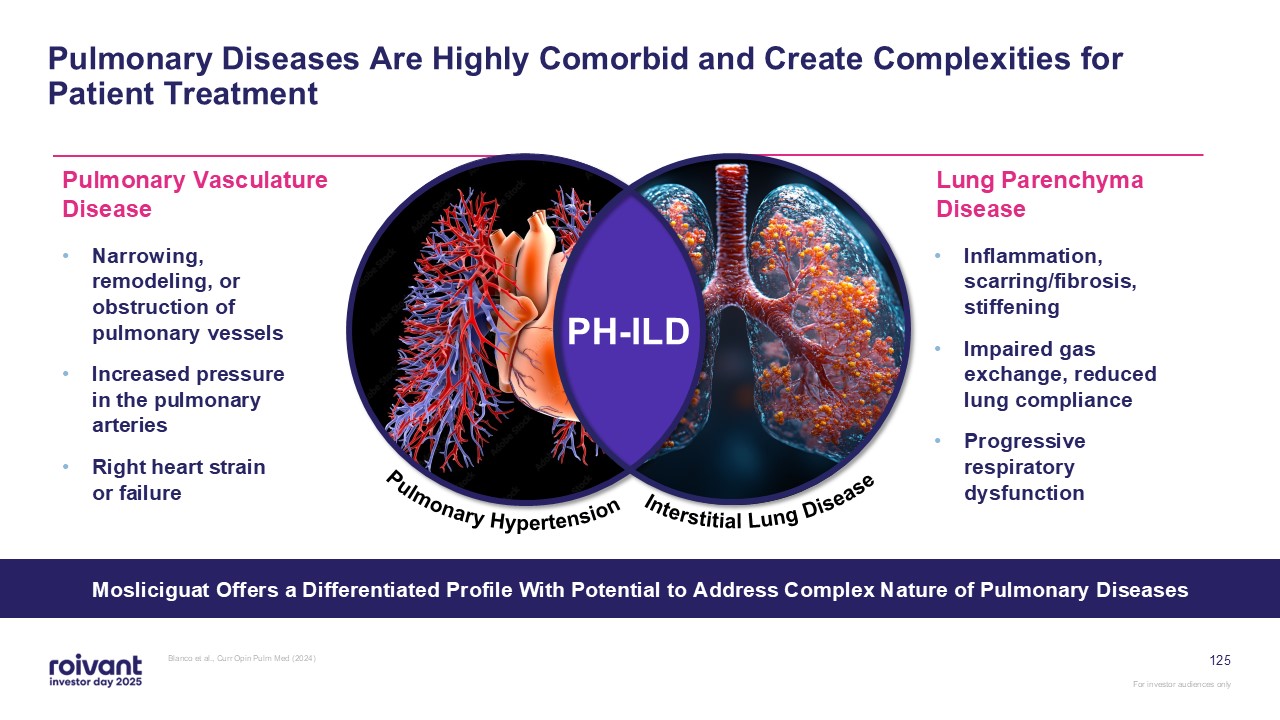
125 Pulmonary Diseases Are Highly Comorbid and Create Complexities for Patient
Treatment Blanco et al., Curr Opin Pulm Med (2024) Mosliciguat Offers a Differentiated Profile With Potential to Address Complex Nature of Pulmonary Diseases Narrowing, remodeling, or obstruction of pulmonary vessels Increased pressure in
the pulmonary arteries Right heart strain or failure Pulmonary Vasculature Disease Inflammation, scarring/fibrosis, stiffening Impaired gas exchange, reduced lung compliance Progressive respiratory dysfunction Lung Parenchyma
Disease PH-ILD Pulmonary Hypertension Interstitial Lung Disease

Mosliciguat Mechanism Video 126

Pulmovant is committed to transforming the lives of patients with pulmonary
diseases For investor audiences only
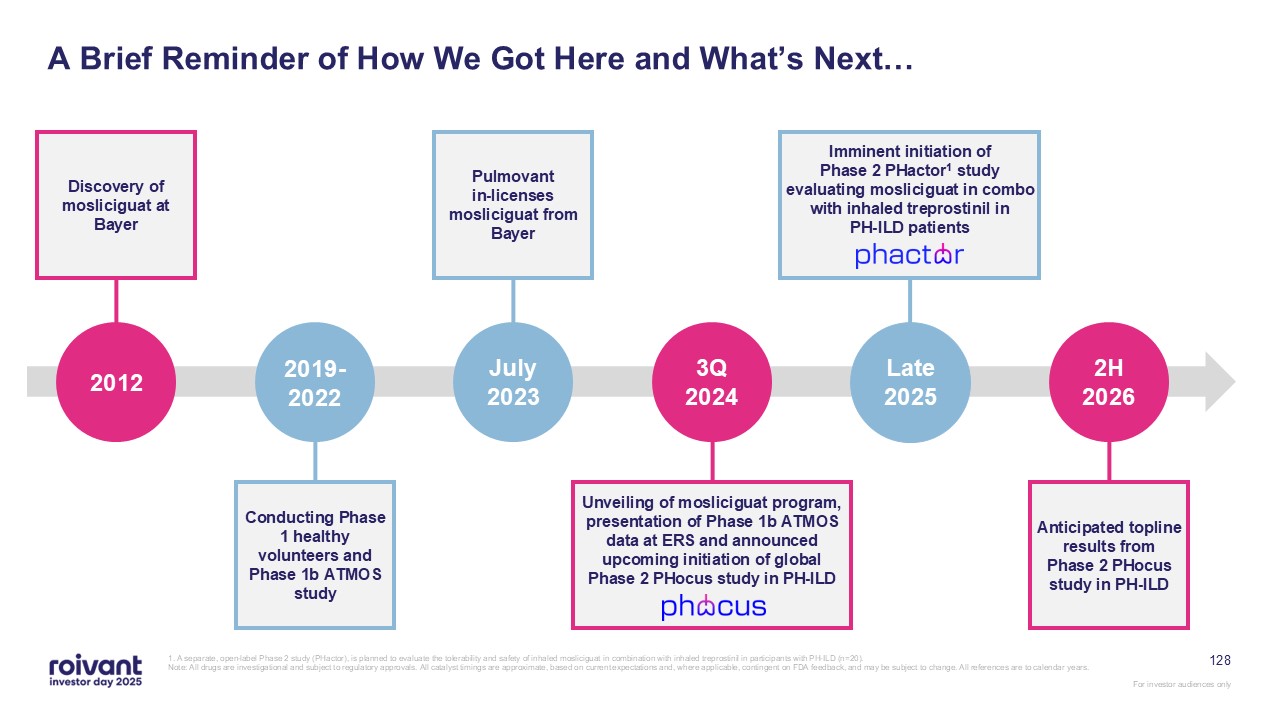
128 A Brief Reminder of How We Got Here and What’s Next… 1. A separate,
open-label Phase 2 study (PHactor), is planned to evaluate the tolerability and safety of inhaled mosliciguat in combination with inhaled treprostinil in participants with PH-ILD (n=20). Note: All drugs are investigational and subject to
regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years. Anticipated topline results from
Phase 2 PHocus study in PH-ILD 2H 2026 Pulmovant in-licenses mosliciguat from Bayer July 2023 Late 2025 Imminent initiation of Phase 2 PHactor1 study evaluating mosliciguat in combo with inhaled treprostinil in PH-ILD patients Discovery
of mosliciguat at Bayer 2012 Unveiling of mosliciguat program, presentation of Phase 1b ATMOS data at ERS and announced upcoming initiation of global Phase 2 PHocus study in PH-ILD 3Q 2024 Conducting Phase 1 healthy volunteers and Phase
1b ATMOS study 2019-2022
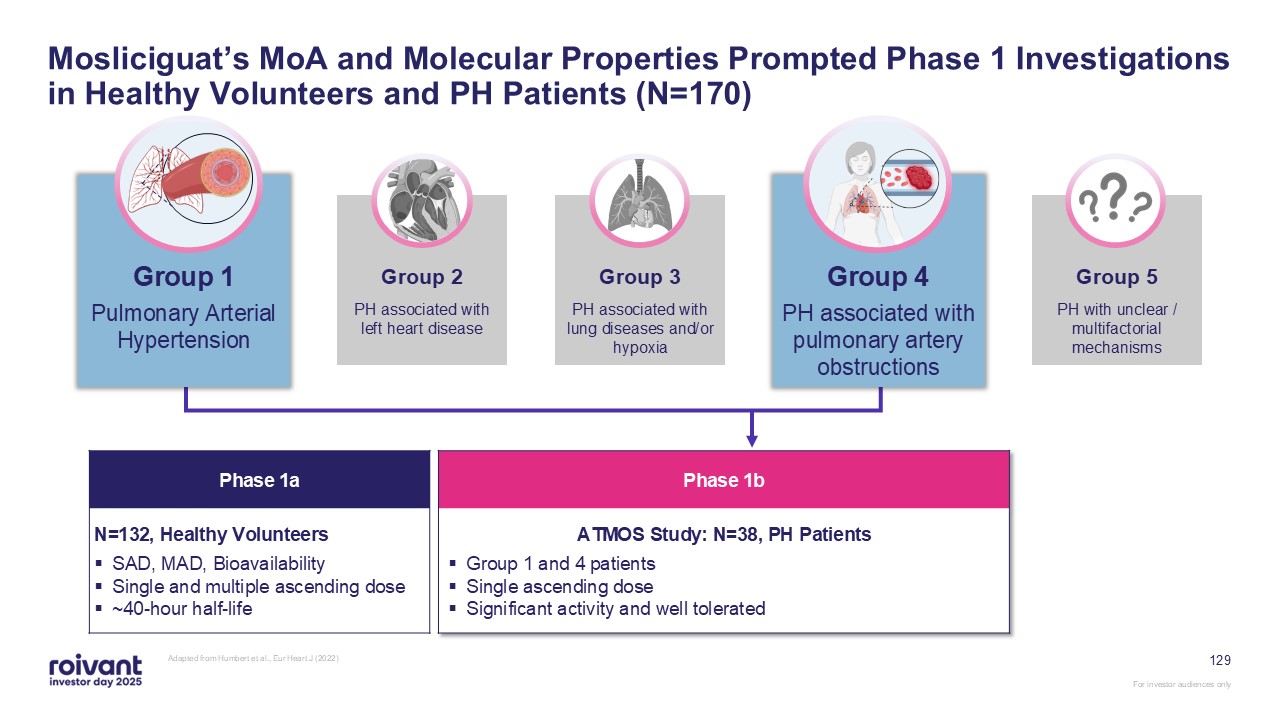
HSF 129 Mosliciguat’s MoA and Molecular Properties Prompted Phase 1
Investigations in Healthy Volunteers and PH Patients (N=170) HSF Group 1 Group 4 Group 2 Group 3 Group 5 Pulmonary Arterial Hypertension PH associated with left heart disease PH associated with lung diseases and/or hypoxia PH
associated with pulmonary artery obstructions PH with unclear / multifactorial mechanisms Phase 1a N=132, Healthy Volunteers SAD, MAD, Bioavailability Single and multiple ascending dose ~40-hour half-life Phase 1b ATMOS Study: N=38,
PH Patients Group 1 and 4 patients Single ascending dose Significant activity and well tolerated Adapted from Humbert et al., Eur Heart J (2022)
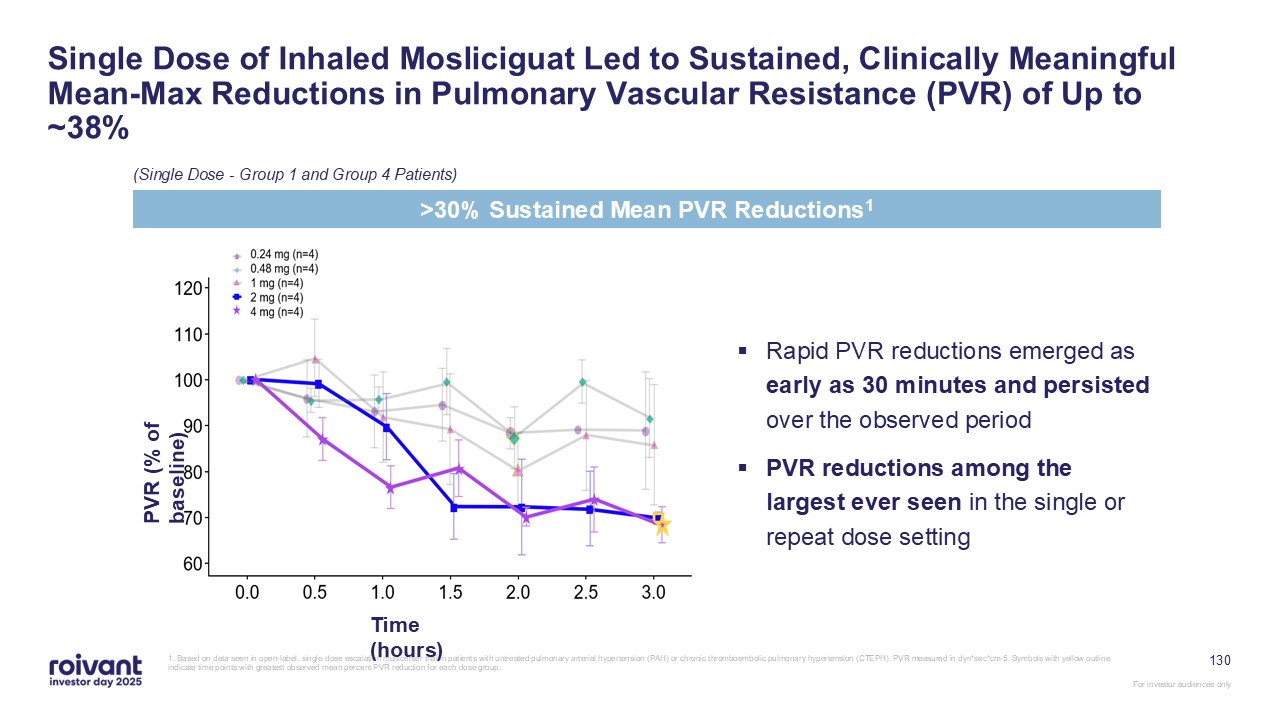
130 (Single Dose - Group 1 and Group 4 Patients) Single Dose of Inhaled
Mosliciguat Led to Sustained, Clinically Meaningful Mean-Max Reductions in Pulmonary Vascular Resistance (PVR) of Up to ~38% 1. Based on data seen in open-label, single-dose escalation multicenter trial in patients with untreated pulmonary
arterial hypertension (PAH) or chronic thromboembolic pulmonary hypertension (CTEPH). PVR measured in dyn*sec*cm-5. Symbols with yellow outline indicate time points with greatest observed mean percent PVR reduction for each dose
group. >30% Sustained Mean PVR Reductions1 Rapid PVR reductions emerged as early as 30 minutes and persisted over the observed period PVR reductions among the largest ever seen in the single or repeat dose setting PVR (% of
baseline) Time (hours)
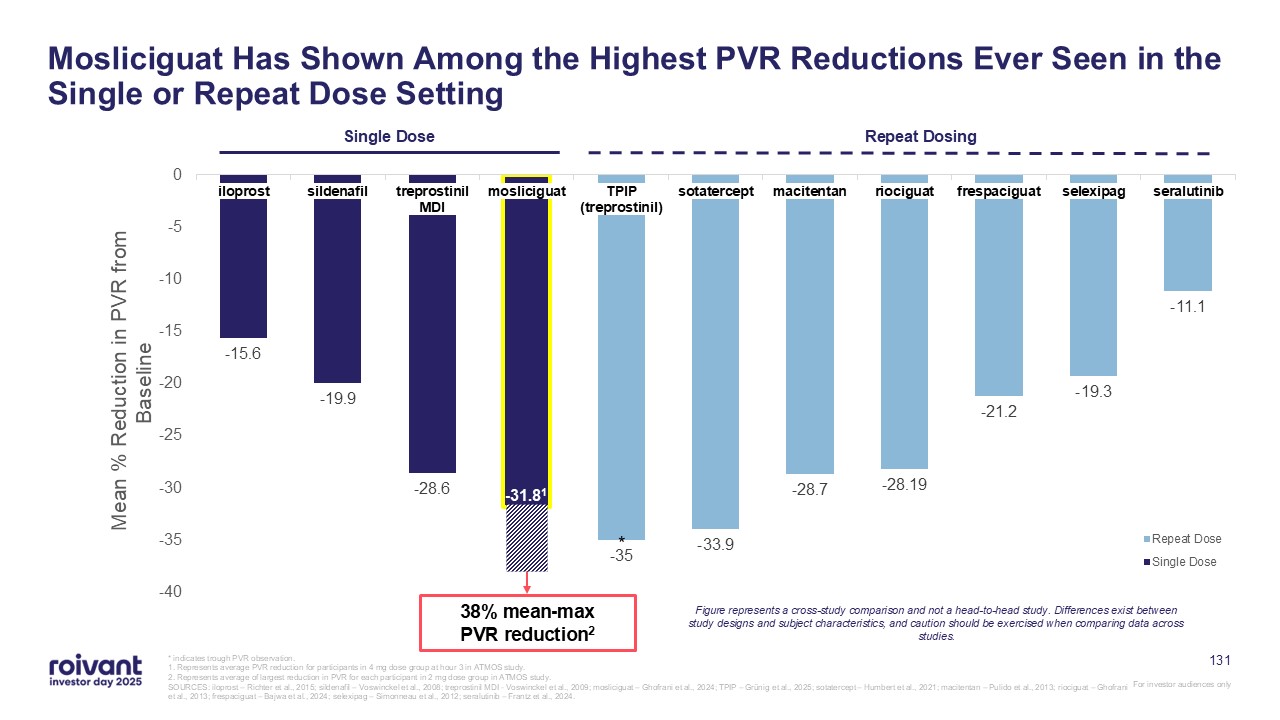
131 Mosliciguat Has Shown Among the Highest PVR Reductions Ever Seen in the
Single or Repeat Dose Setting * -31.81 * indicates trough PVR observation. 1. Represents average PVR reduction for participants in 4 mg dose group at hour 3 in ATMOS study. 2. Represents average of largest reduction in PVR for each
participant in 2 mg dose group in ATMOS study. SOURCES: iloprost – Richter et al., 2015; sildenafil – Voswinckel et al., 2008; treprostinil MDI - Voswinckel et al., 2009; mosliciguat – Ghofrani et al., 2024; TPIP – Grünig et al., 2025;
sotatercept – Humbert et al., 2021; macitentan – Pulido et al., 2013; riociguat – Ghofrani et al., 2013; frespaciguat – Bajwa et al., 2024; selexipag – Simonneau et al., 2012; seralutinib – Frantz et al., 2024. Figure represents a
cross-study comparison and not a head-to-head study. Differences exist between study designs and subject characteristics, and caution should be exercised when comparing data across studies. Single Dose Repeat Dosing 38% mean-max PVR
reduction2 * -31.81
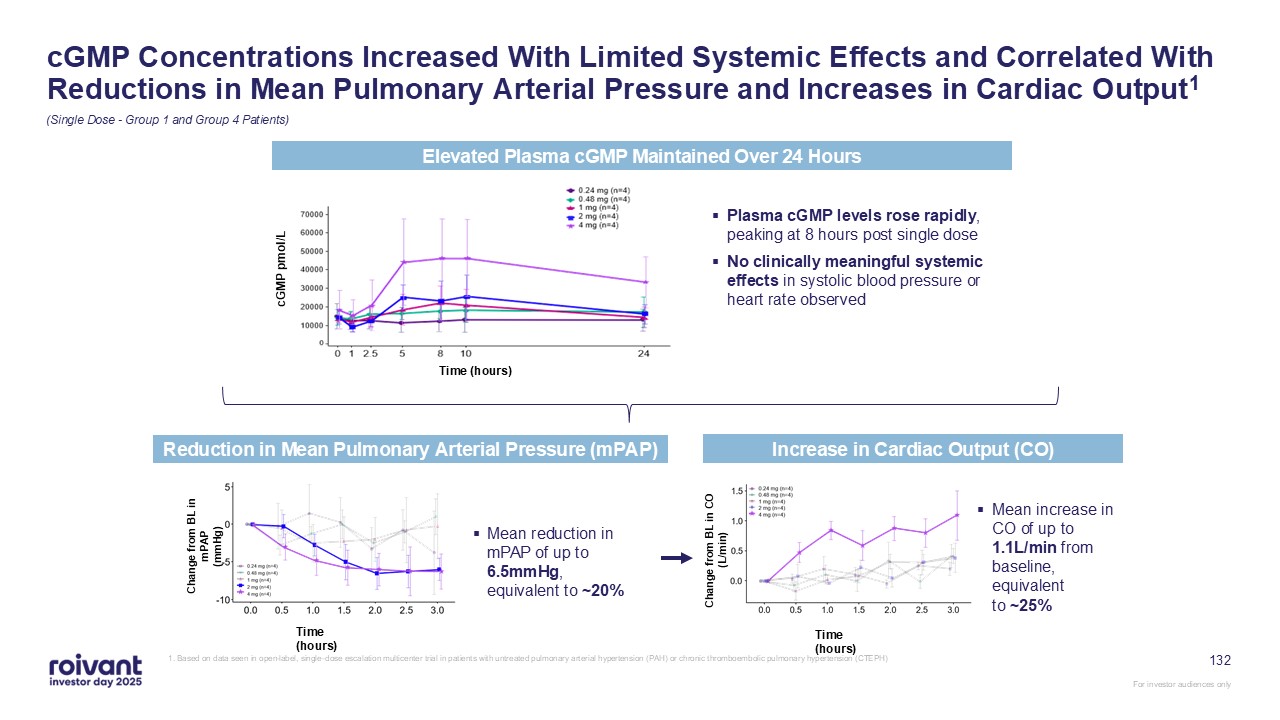
132 (Single Dose - Group 1 and Group 4 Patients) cGMP Concentrations Increased
With Limited Systemic Effects and Correlated With Reductions in Mean Pulmonary Arterial Pressure and Increases in Cardiac Output1 Increase in Cardiac Output (CO) Mean increase in CO of up to 1.1L/min from baseline, equivalent to
~25% Change from BL in CO (L/min) Time (hours) Reduction in Mean Pulmonary Arterial Pressure (mPAP) Mean reduction in mPAP of up to 6.5mmHg, equivalent to ~20% Change from BL in mPAP (mmHg) Time (hours) Elevated Plasma cGMP Maintained
Over 24 Hours Plasma cGMP levels rose rapidly, peaking at 8 hours post single dose No clinically meaningful systemic effects in systolic blood pressure or heart rate observed cGMP pmol/L Time (hours) 1. Based on data seen in open-label,
single-dose escalation multicenter trial in patients with untreated pulmonary arterial hypertension (PAH) or chronic thromboembolic pulmonary hypertension (CTEPH)
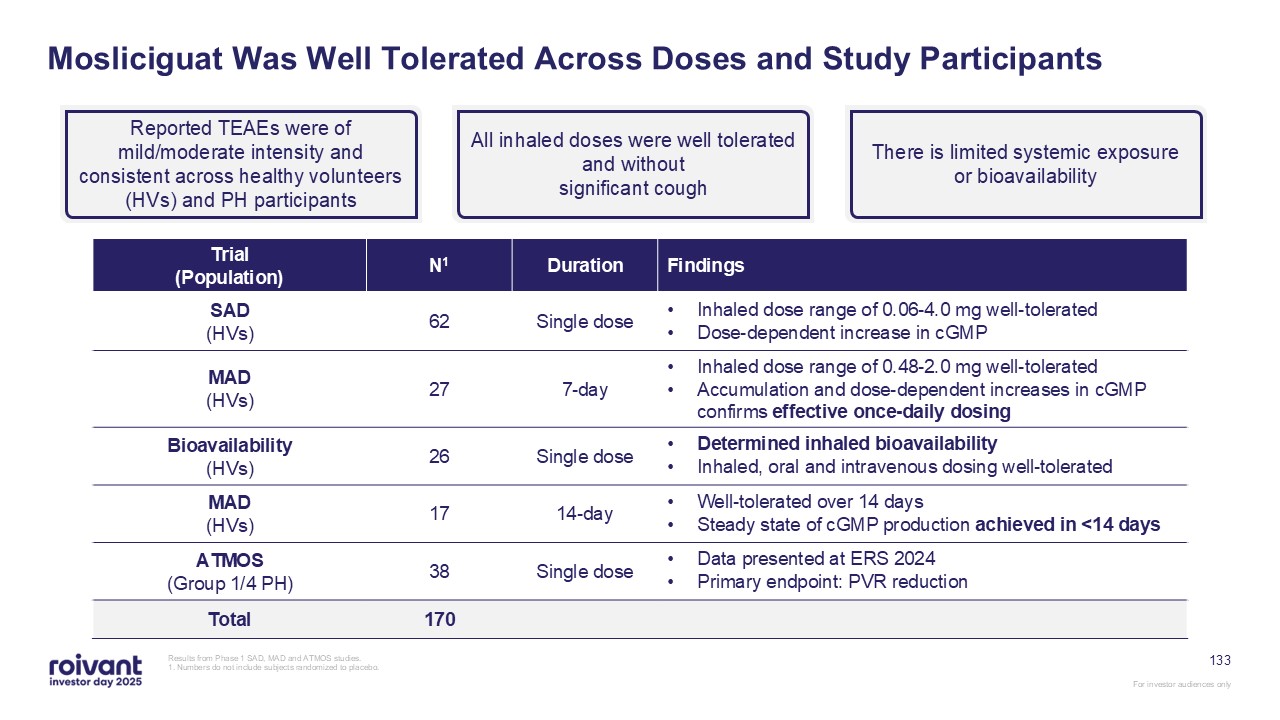
133 Mosliciguat Was Well Tolerated Across Doses and Study Participants Results
from Phase 1 SAD, MAD and ATMOS studies. 1. Numbers do not include subjects randomized to placebo. Reported TEAEs were of mild/moderate intensity and consistent across healthy volunteers (HVs) and PH participants All inhaled doses were
well tolerated and without significant cough There is limited systemic exposure or bioavailability Trial(Population) N1 Duration Findings SAD(HVs) 62 Single dose Inhaled dose range of 0.06-4.0 mg well-tolerated Dose-dependent
increase in cGMP MAD(HVs) 27 7-day Inhaled dose range of 0.48-2.0 mg well-tolerated Accumulation and dose-dependent increases in cGMP confirms effective once-daily dosing Bioavailability(HVs) 26 Single dose Determined inhaled
bioavailability Inhaled, oral and intravenous dosing well-tolerated MAD(HVs) 17 14-day Well-tolerated over 14 days Steady state of cGMP production achieved in <14 days ATMOS(Group 1/4 PH) 38 Single dose Data presented at ERS
2024 Primary endpoint: PVR reduction Total 170

134 Phase 1 and ATMOS Demonstrated Mosliciguat Has the Attributes to
Potentially Address Complex, Heterogeneous Pulmonary Disorders Like PH-ILD 1. Evidenced by data demonstrated in ATMOS and Phase 1 studies conducted in healthy volunteers 2. Becker-Peslster et al., Respir Res
(2022) PH-ILD MOSLICIGUAT Lung is the primary site of the disease Target delivery to the lungs with deep lung deposition1 High dosing burden with multiple daily inhalations Convenient once-daily dosing Current therapies are poorly
tolerated and can increase cough Well-tolerated, with limited cough and systemic side effects1 Interplay of vascular remodeling and parenchymal scarring Promotes vasodilation1,2 and may exert antifibrotic and anti-inflammatory effects2
1x day cGMP
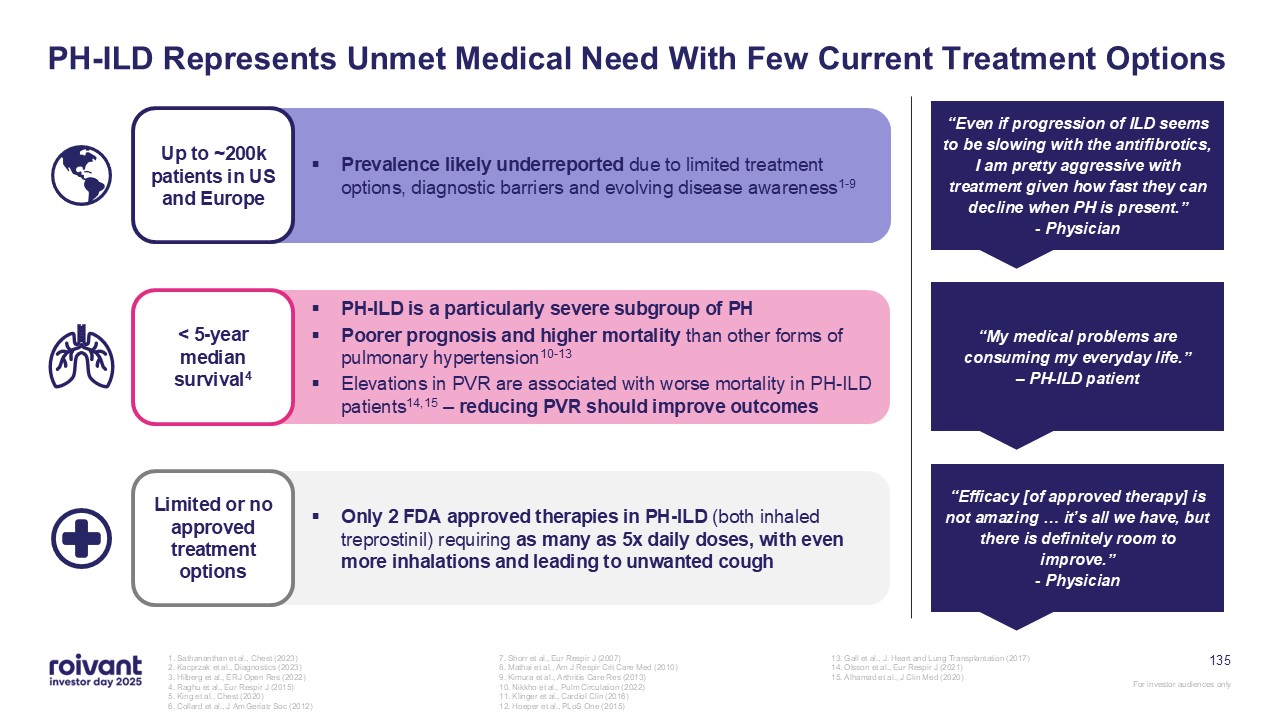
135 PH-ILD Represents Unmet Medical Need With Few Current Treatment Options 1.
Sathananthan et al., Chest (2023) 2. Kacprzak et al., Diagnostics (2023) 3. Hilberg et al., ERJ Open Res (2022) 4. Raghu et al., Eur Respir J (2015) 5. King et al., Chest (2020) 6. Collard et al., J Am Geriatr Soc (2012) 7. Shorr et
al., Eur Respir J (2007) 8. Mathai et al., Am J Respir Crit Care Med (2010) 9. Kimura et al., Arthritis Care Res (2013) 10. Nikkho et al., Pulm Circulation (2022) 11. Klinger et al., Cardiol Clin (2016) 12. Hoeper et al., PLoS One
(2015) 13. Gall et al., J. Heart and Lung Transplantation (2017) 14. Olsson et al., Eur Respir J (2021) 15. Alhamad et al., J Clin Med (2020) “Even if progression of ILD seems to be slowing with the antifibrotics, I am pretty aggressive
with treatment given how fast they can decline when PH is present.” - Physician “My medical problems are consuming my everyday life.” – PH-ILD patient “Efficacy [of approved therapy] is not amazing … it’s all we have, but there is
definitely room to improve.” - Physician Limited or no approved treatment options Only 2 FDA approved therapies in PH-ILD (both inhaled treprostinil) requiring as many as 5x daily doses, with even more inhalations and leading to unwanted
cough Up to ~200k patients in US and Europe Prevalence likely underreported due to limited treatment options, diagnostic barriers and evolving disease awareness1-9 < 5-year median survival4 PH-ILD is a particularly severe subgroup of
PH Poorer prognosis and higher mortality than other forms of pulmonary hypertension10-13 Elevations in PVR are associated with worse mortality in PH-ILD patients14,15 – reducing PVR should improve outcomes
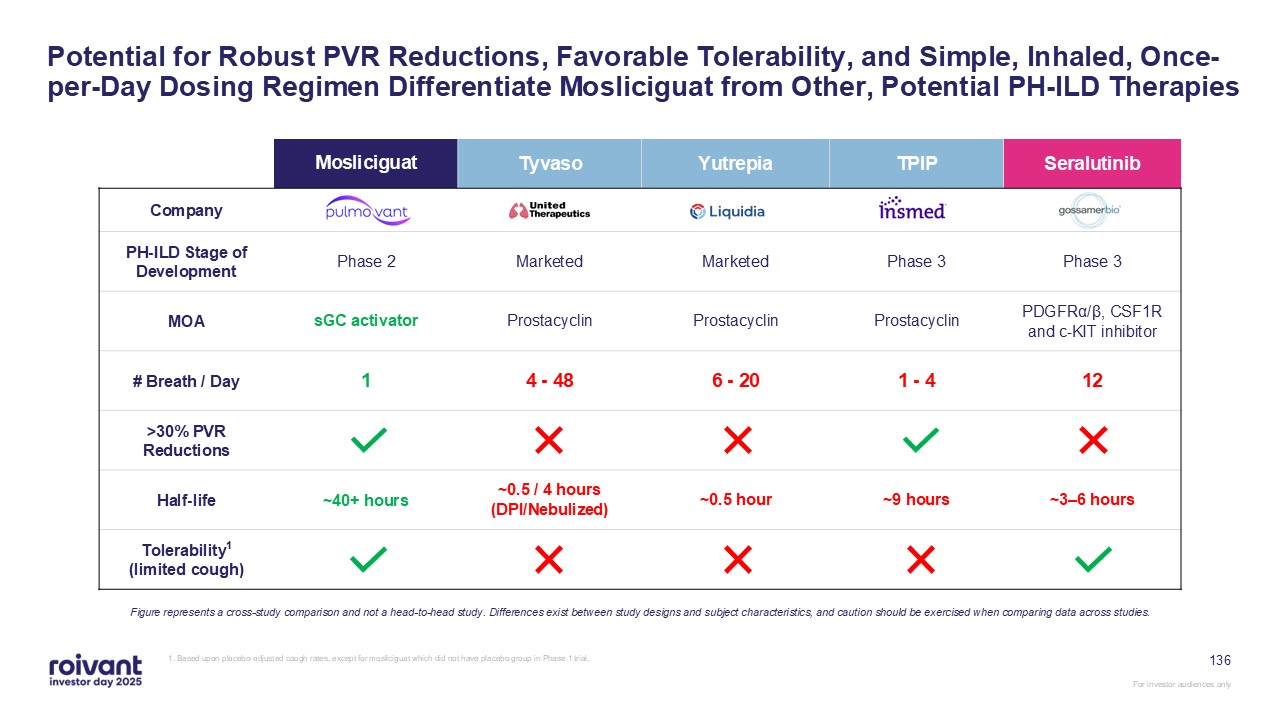
136 Potential for Robust PVR Reductions, Favorable Tolerability, and Simple,
Inhaled, Once-per-Day Dosing Regimen Differentiate Mosliciguat from Other, Potential PH-ILD Therapies Figure represents a cross-study comparison and not a head-to-head study. Differences exist between study designs and subject
characteristics, and caution should be exercised when comparing data across studies. Mosliciguat Tyvaso Yutrepia TPIP Seralutinib Company PH-ILD Stage of Development Phase 2 Marketed Marketed Phase 3 Phase 3 MOA sGC
activator Prostacyclin Prostacyclin Prostacyclin PDGFRα/β, CSF1R and c-KIT inhibitor # Breath / Day 1 4 - 48 6 - 20 1 - 4 12 >30% PVR Reductions Half-life ~40+ hours ~0.5 / 4 hours (DPI/Nebulized) ~0.5 hour ~9 hours ~3–6
hours Tolerability1 (limited cough) 1. Based upon placebo-adjusted cough rates, except for mosliciguat which did not have placebo group in Phase 1 trial.
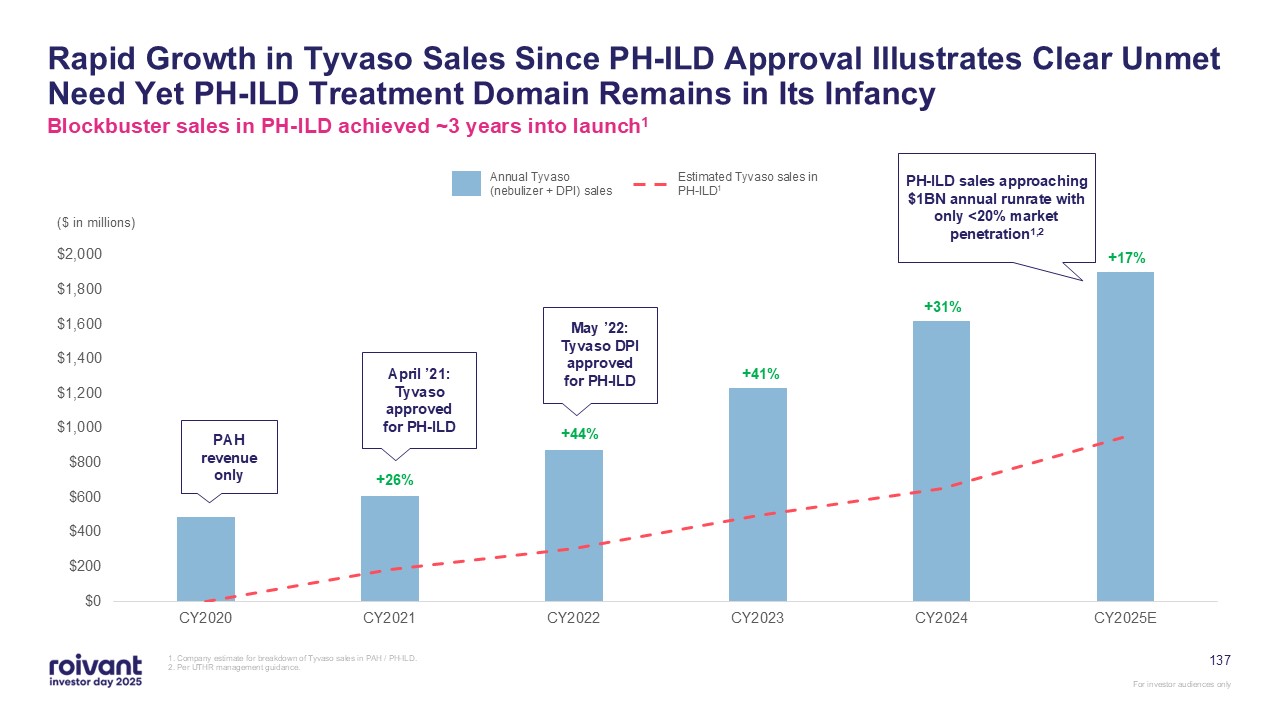
137 Rapid Growth in Tyvaso Sales Since PH-ILD Approval Illustrates Clear Unmet
Need Yet PH-ILD Treatment Domain Remains in Its Infancy Blockbuster sales in PH-ILD achieved ~3 years into launch1 1. Company estimate for breakdown of Tyvaso sales in PAH / PH-ILD.2. Per UTHR management guidance. April ’21: Tyvaso
approved for PH-ILD ($ in millions) +26% +44% +41% Annual Tyvaso (nebulizer + DPI) sales Estimated Tyvaso sales in PH-ILD1 +31% +17% PH-ILD sales approaching $1BN annual runrate with only <20% market penetration1,2 May ’22:
Tyvaso DPI approved for PH-ILD PAH revenue only
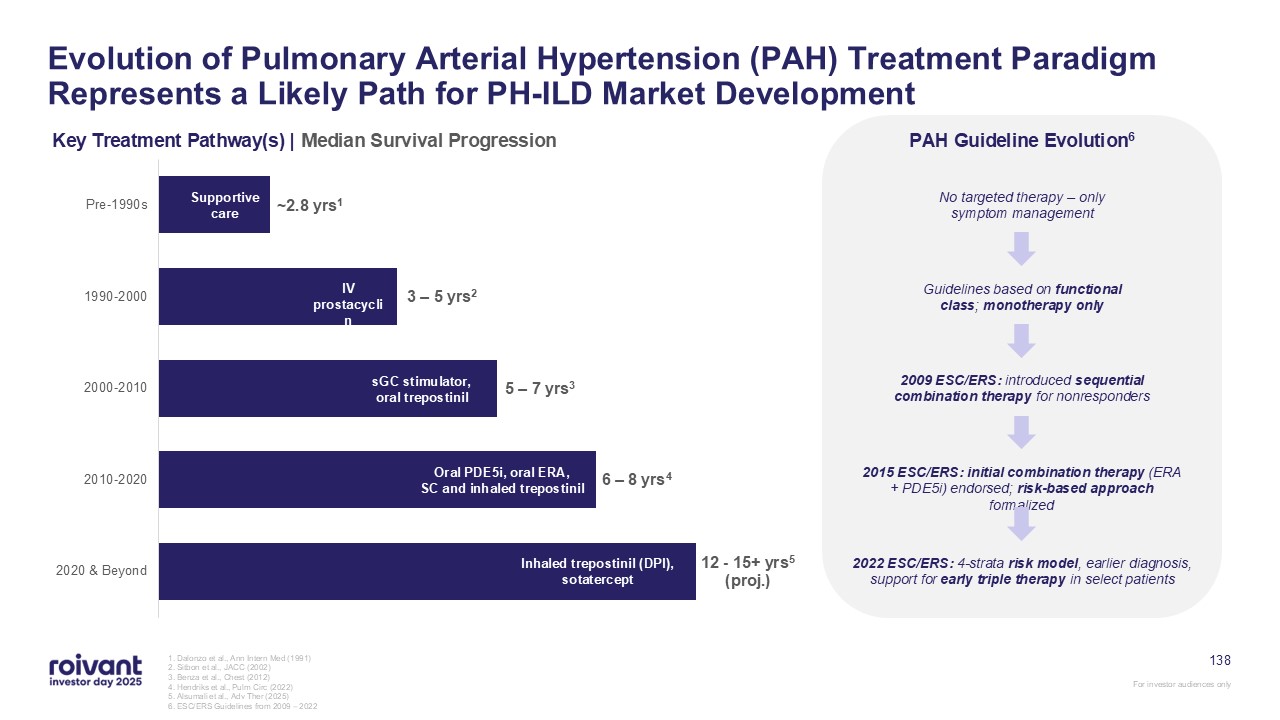
138 Evolution of Pulmonary Arterial Hypertension (PAH) Treatment Paradigm
Represents a Likely Path for PH-ILD Market Development Key Treatment Pathway(s) | Median Survival Progression 1. Dalonzo et al., Ann Intern Med (1991) 2. Sitbon et al., JACC (2002) 3. Benza et al., Chest (2012) 4. Hendriks et al., Pulm
Circ (2022) 5. Alsumali et al., Adv Ther (2025) 6. ESC/ERS Guidelines from 2009 – 2022 Supportive care IV prostacyclin sGC stimulator, oral trepostinil Oral PDE5i, oral ERA, SC and inhaled trepostinil Inhaled trepostinil (DPI),
sotatercept PAH Guideline Evolution6 No targeted therapy – only symptom management Guidelines based on functional class; monotherapy only 2009 ESC/ERS: introduced sequential combination therapy for nonresponders 2015 ESC/ERS: initial
combination therapy (ERA + PDE5i) endorsed; risk-based approach formalized 2022 ESC/ERS: 4-strata risk model, earlier diagnosis, support for early triple therapy in select patients
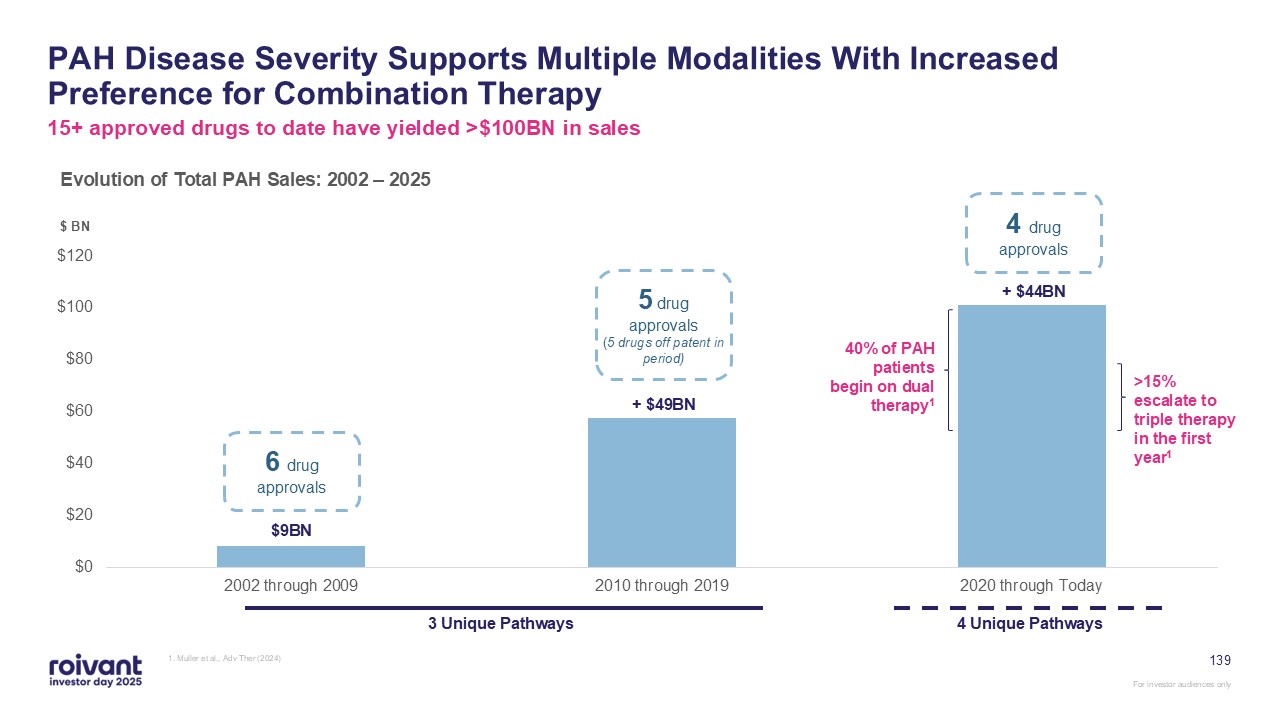
139 PAH Disease Severity Supports Multiple Modalities With Increased Preference
for Combination Therapy 1. Muller et al., Adv Ther (2024) Evolution of Total PAH Sales: 2002 – 2025 $9BN + $49BN + $44BN 6 drug approvals 5 drug approvals(5 drugs off patent in period) 4 drug approvals $ BN 15+ approved drugs to
date have yielded >$100BN in sales 3 Unique Pathways 4 Unique Pathways 40% of PAH patients begin on dual therapy1 >15% escalate to triple therapy in the first year1
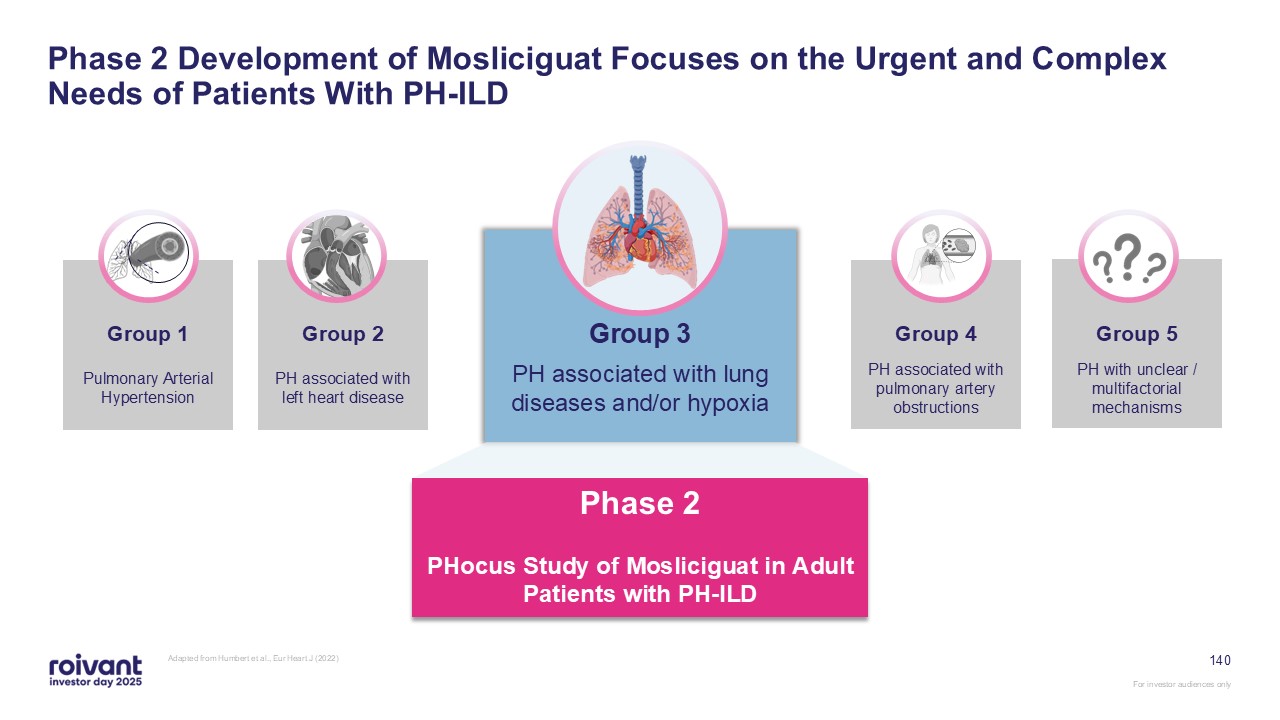
140 Phase 2 Development of Mosliciguat Focuses on the Urgent and Complex Needs
of Patients With PH-ILD Adapted from Humbert et al., Eur Heart J (2022) Group 3 PH associated with lung diseases and/or hypoxia Phase 2 PHocus Study of Mosliciguat in Adult Patients with PH-ILD PH associated with left heart disease PH
associated with pulmonary artery obstructions PH with unclear / multifactorial mechanisms Group 2 Group 1 Group 4 Group 5 Pulmonary Arterial Hypertension
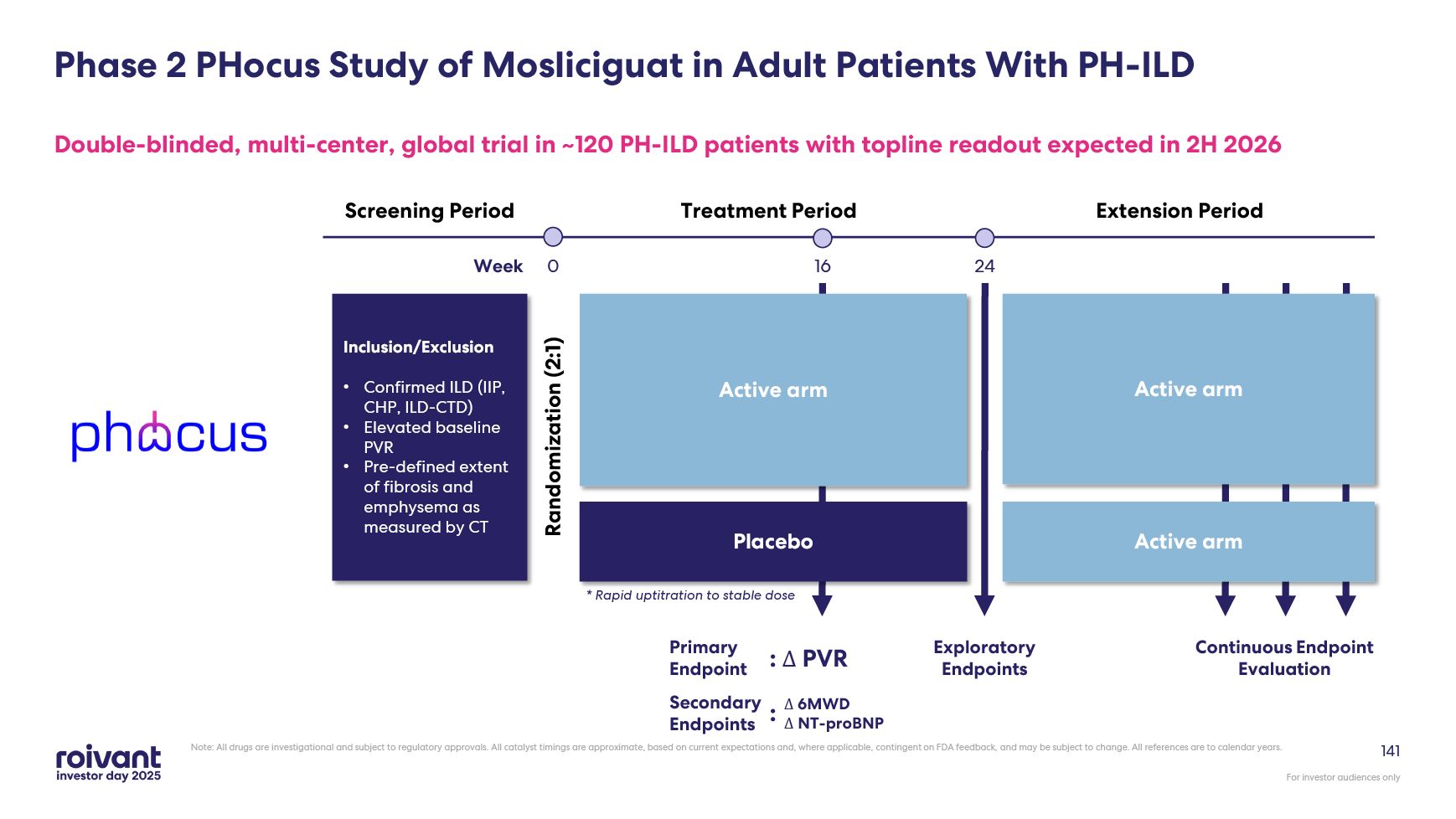
141 Phase 2 PHocus Study of Mosliciguat in Adult Patients With
PH-ILD Double-blinded, multi-center, global trial in ~120 PH-ILD patients with topline readout expected in 2H 2026 Extension Period Treatment Period Screening Period Inclusion/Exclusion Confirmed ILD (IIP, CHP, ILD-CTD) Elevated
baseline PVR Pre-defined extent of fibrosis and emphysema as measured by CT Randomization (2:1) Week 0 16 24 Exploratory Endpoints Placebo Active arm Primary Endpoint : Δ PVR Secondary Endpoints Δ 6MWDΔ NT-proBNP : * Rapid
uptitration to stable dose Active arm Active arm Continuous Endpoint Evaluation Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where
applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years.

Phase 2 PHactor Study of Mosliciguat in Combination with Inhaled Trepostinil A
separate, open-label Phase 2 study is planned to evaluate the tolerability and safety of inhaled mosliciguat in combination with inhaled treprostinil in participants with PH-ILD (n=20). This study is expected to initiate imminently. Note:
All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to
calendar years. 142

143 In Summary: Mosliciguat Among the best PVR reductions seen to date with
differentiated MoA, convenient once-daily dosing and favorable safety profile PH-ILD is an area of unmet medical need – tractable market with only one approved mechanism Topline data from ongoing Phase 2 study (PHocus) expected in 2H 2026;
if successful, has potential to define standard of care in PH-ILD Mosliciguat will potentially represent the first non-treprostinil treatment option for PH-ILD patients Note: All drugs are investigational and subject to regulatory
approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years. For investor audiences only
Q&A

145 Genevant & Arbutus LNP Litigation Lindsay Androski Special Counsel,
Genevant CEO, Arbutus Matt Gline CEO, Roivant

146 We believe that both the Moderna COVID-19 vaccine (SPIKEVAX) and
Pfizer/BioNTech’s COVID-19 vaccine (COMIRNATY) infringe multiple Genevant/Arbutus LNP patents In the ex-US Moderna litigation, initial court hearings and rulings are expected in 2026 Markman rulings (claim construction) have been issued in
both US cases – viewed by Genevant generally to be favorable Global COVID-19 vaccine sales since launch have been ~$145BN between Moderna and Pfizer/BioNTech In the US Moderna litigation, a jury trial has been scheduled for March 2026.
Awaiting court scheduling in the Pfizer/BioNTech litigation Key Takeaways: LNP Litigation For investor audiences only Note: All references are to calendar years and are approximate and subject to change. The timing of the
litigation-related events noted above is subject to change, including at the discretion of the court. See Slide 2 for further information on these forward-looking statements
Genevant and Arbutus Corporate History 2006 Protiva acquired Tekmira, another
company working on LNP technology Roivant set out to assemble a leading global HBV company by merging OnCore Biopharma with Protiva/Tekmira to form Arbutus Biopharma Roivant and Arbutus launched Genevant as a joint venture to focus on
Arbutus’ LNP and ligand conjugate delivery technologies, with Arbutus’ focus remaining on HBV therapeutics 2008 2015 2018 Protiva and Alnylam (an RNAi developer) published a landmark study in Nature demonstrating the first effective gene
silencing in monkeys, using Protiva’s LNP technology 2000 Ian MacLachlan co-founded Protiva, which became a pioneer in LNP technology 147

148 A Leading Nucleic Acid Delivery Company Industry-leading LNP delivery
capabilities and IP portfolio Selective collaboration business model, partnering with payload companies to develop innovative nucleic acid medicines The first LNP technology to be part of an FDA-approved RNA product, Alnylam’s Onpattro®
developed under LNP license from Arbutus
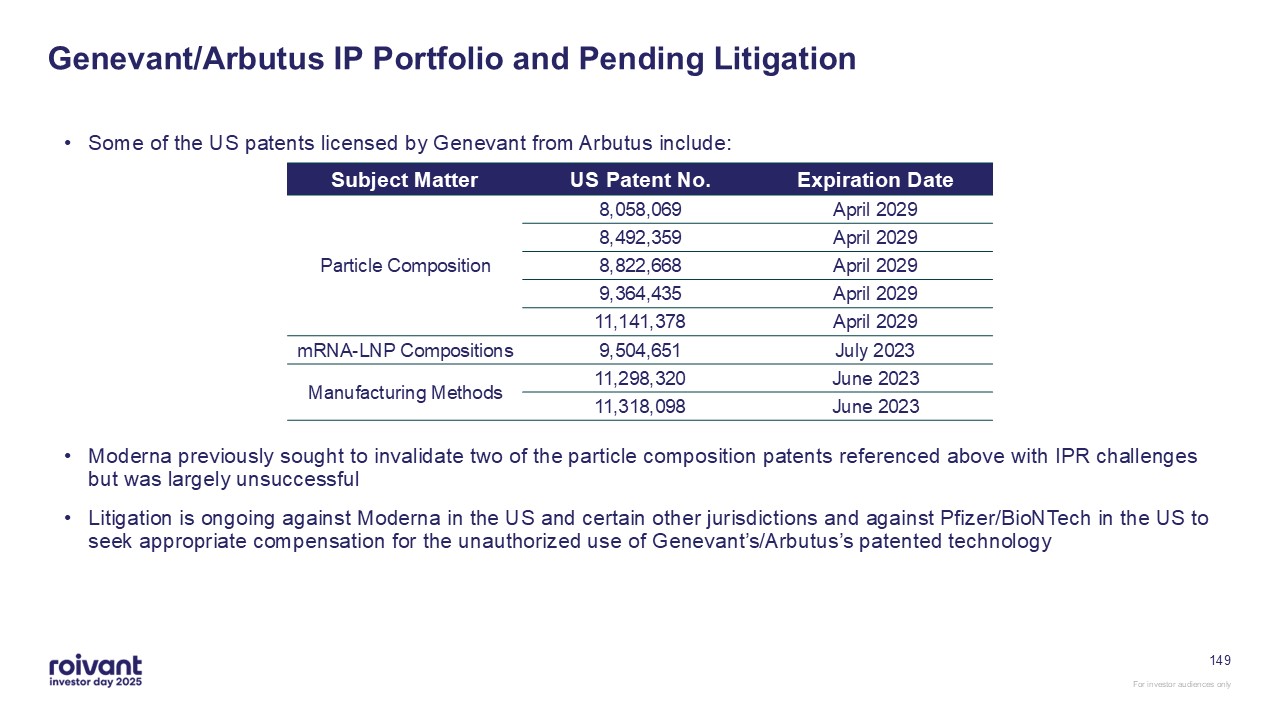
Some of the US patents licensed by Genevant from Arbutus include: Moderna
previously sought to invalidate two of the particle composition patents referenced above with IPR challenges but was largely unsuccessful Litigation is ongoing against Moderna in the US and certain other jurisdictions and against
Pfizer/BioNTech in the US to seek appropriate compensation for the unauthorized use of Genevant’s/Arbutus’s patented technology 149 Genevant/Arbutus IP Portfolio and Pending Litigation Subject Matter US Patent No. Expiration
Date Particle Composition 8,058,069 April 2029 8,492,359 April 2029 8,822,668 April 2029 9,364,435 April 2029 11,141,378 April 2029 mRNA-LNP Compositions 9,504,651 July 2023 Manufacturing Methods 11,298,320 June
2023 11,318,098 June 2023
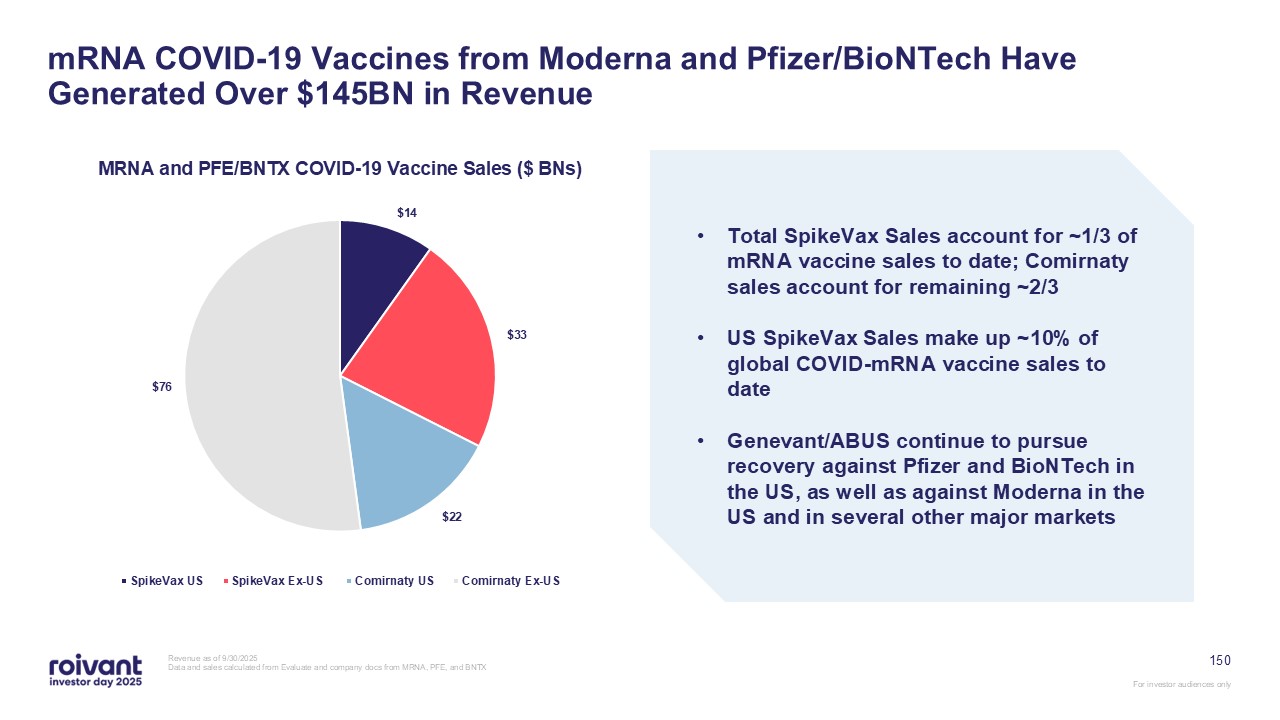
150 mRNA COVID-19 Vaccines from Moderna and Pfizer/BioNTech Have Generated Over
$145BN in Revenue Revenue as of 9/30/2025 Data and sales calculated from Evaluate and company docs from MRNA, PFE, and BNTX Total SpikeVax Sales account for ~1/3 of mRNA vaccine sales to date; Comirnaty sales account for remaining
~2/3 US SpikeVax Sales make up ~10% of global COVID-mRNA vaccine sales to date Genevant/ABUS continue to pursue recovery against Pfizer and BioNTech in the US, as well as against Moderna in the US and in several other major markets

Moderna Case

152 In February 2022, Genevant and Arbutus Jointly Filed a Complaint Against
Moderna Asserting Patent Infringement on Patents Related to LNP Technology Case filed in the US District Court for the District of Delaware asserting infringement of six patents Genevant and Arbutus did not seek an injunction or otherwise
to impede the sale, manufacture, or distribution of Moderna’s COVID-19 vaccine, given the unprecedented global emergency We recognize the important work of Moderna that helped lead to a lifesaving vaccine in record time That success was
built on, and made possible by, the substantial advances and contributions of Arbutus and Genevant scientists, which Moderna utilized extensively well before COVID-19 The filing of this lawsuit was necessary because Moderna did not pursue
and obtain a license to Genevant’s LNP technology for COVID-19
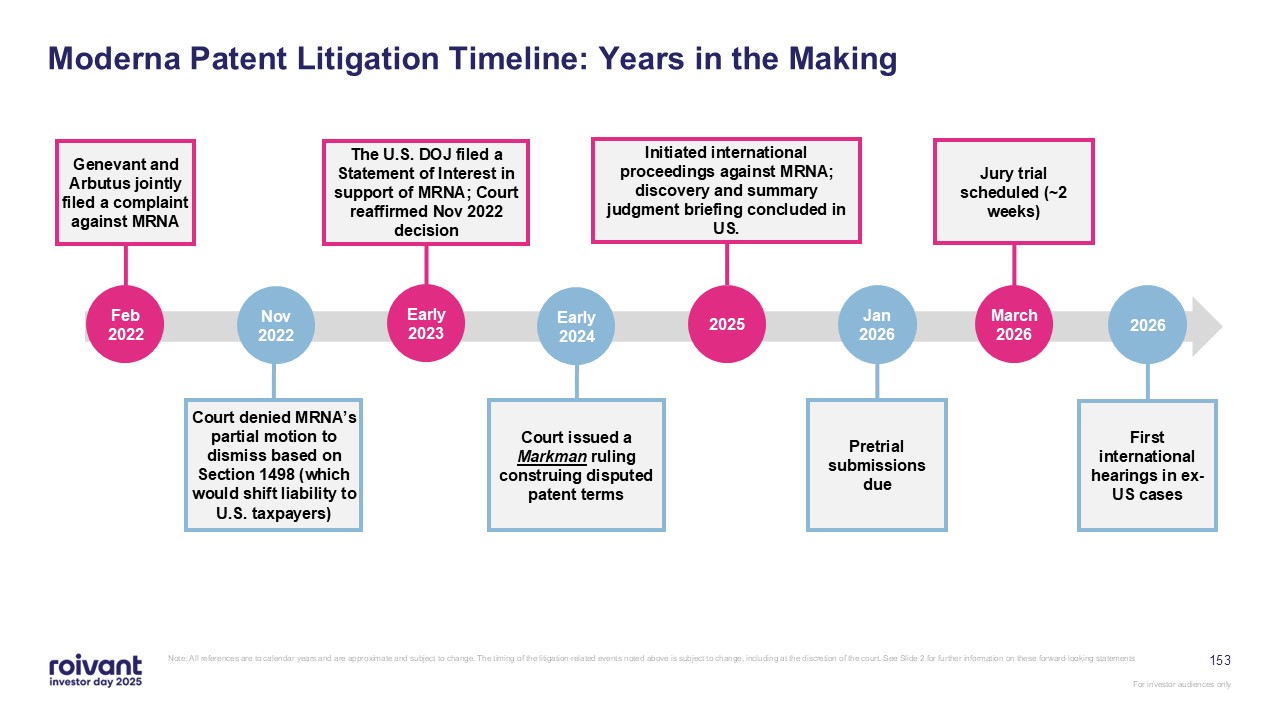
153 Moderna Patent Litigation Timeline: Years in the Making Note: All
references are to calendar years and are approximate and subject to change. The timing of the litigation-related events noted above is subject to change, including at the discretion of the court. See Slide 2 for further information on these
forward-looking statements Early 2023 The U.S. DOJ filed a Statement of Interest in support of MRNA; Court reaffirmed Nov 2022 decision Feb 2022 Genevant and Arbutus jointly filed a complaint against MRNA 2025 Initiated international
proceedings against MRNA; discovery and summary judgment briefing concluded in US. Jan 2026 Pretrial submissions due Early 2024 Court issued a Markman ruling construing disputed patent terms Nov 2022 Court denied MRNA’s partial motion
to dismiss based on Section 1498 (which would shift liability to U.S. taxpayers) March 2026 Jury trial scheduled (~2 weeks) 2026 First international hearings in ex-US cases
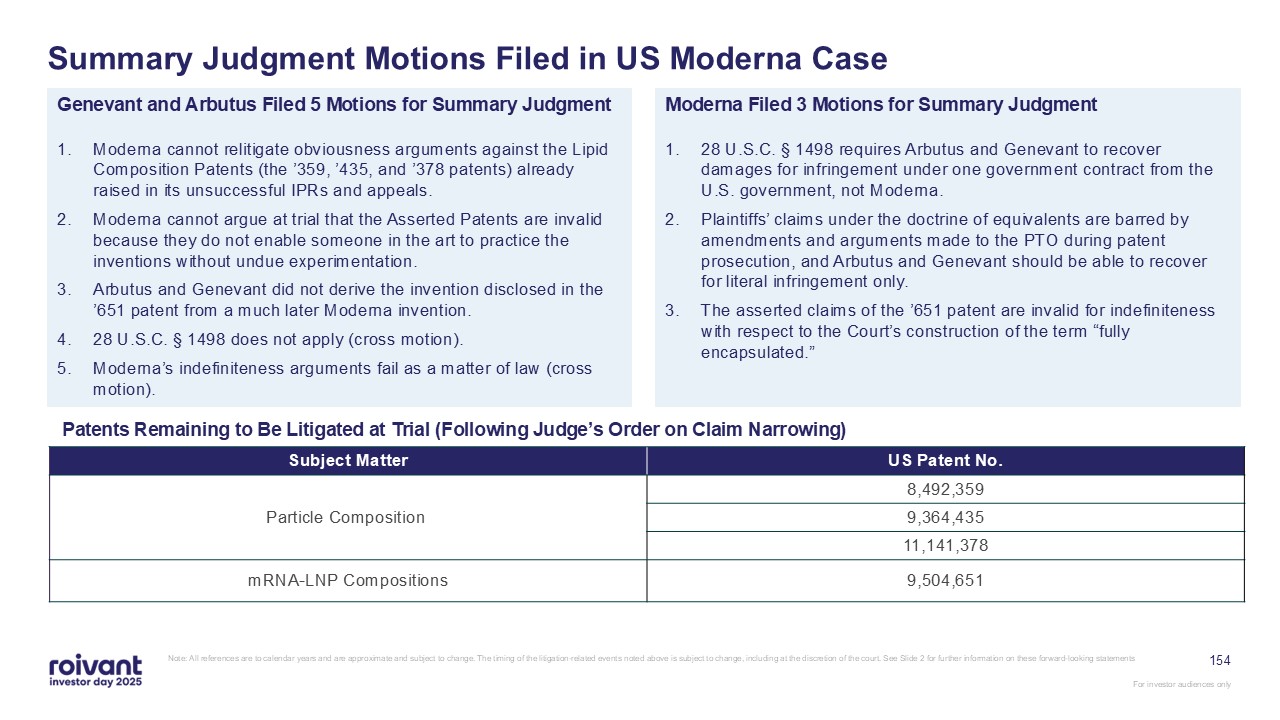
154 Summary Judgment Motions Filed in US Moderna Case Note: All references are
to calendar years and are approximate and subject to change. The timing of the litigation-related events noted above is subject to change, including at the discretion of the court. See Slide 2 for further information on these forward-looking
statements Genevant and Arbutus Filed 5 Motions for Summary Judgment Moderna cannot relitigate obviousness arguments against the Lipid Composition Patents (the ’359, ’435, and ’378 patents) already raised in its unsuccessful IPRs and
appeals. Moderna cannot argue at trial that the Asserted Patents are invalid because they do not enable someone in the art to practice the inventions without undue experimentation. Arbutus and Genevant did not derive the invention disclosed
in the ’651 patent from a much later Moderna invention. 28 U.S.C. § 1498 does not apply (cross motion). Moderna’s indefiniteness arguments fail as a matter of law (cross motion). Moderna Filed 3 Motions for Summary Judgment 28 U.S.C. §
1498 requires Arbutus and Genevant to recover damages for infringement under one government contract from the U.S. government, not Moderna. Plaintiffs’ claims under the doctrine of equivalents are barred by amendments and arguments made to
the PTO during patent prosecution, and Arbutus and Genevant should be able to recover for literal infringement only. The asserted claims of the ’651 patent are invalid for indefiniteness with respect to the Court’s construction of the term
“fully encapsulated.” Patents Remaining to Be Litigated at Trial (Following Judge’s Order on Claim Narrowing) Subject Matter US Patent No. Particle Composition 8,492,359 9,364,435 11,141,378 mRNA-LNP Compositions 9,504,651

155 Moderna’s Motions to Exclude Genevant/Arbutus’ Expert Testimony Damages
methodologies Opinions regarding willful infringement Fractionation testing Opinions regarding infringement Opinions regarding applicability of Section 1498 Genevant/Arbutus’ Motions to Exclude Moderna’s Expert Testimony Damages
methodologies Certain opinions regarding Infringement, Doctrine of Equivalents, Written Description and Enablement Opinions regarding obviousness Daubert Motions: Judge to Decide Admissibility of Certain Expert Testimony

Key Upcoming Milestones in US Litigation Judge to rule on summary judgment
motions, including decision on § 1498, before the jury trial in March Judge will rule on Daubert motions, which could narrow the expert testimony allowed to be presented on both sides or impact presentations on damages, infringement, willful
infringement, and invalidity A jury trial is currently scheduled to start on March 9, 2026, in the U.S. District Court of Delaware. Jury selection will occur that day Genevant and Arbutus first, and Moderna second, will present their cases
over the course of ~2 weeks Jury will deliberate, and parties will remain in Delaware until the jury issues their verdict Jury Trial Summary Judgement/Daubert Motions Any jury verdict is expected to be appealed Post-trial motions
entertained by trial court in period after trial An appeal could take an additional 18-24 months If the jury rules favorably for Genevant/Arbutus, to obtain a stay of execution of judgment pending appeal, Moderna would likely need to
obtain a bond or post cash collateral with the court within 30 days After the Trial 156 Note: All references are to calendar years and are approximate and subject to change. The timing of the litigation-related events noted above is
subject to change, including at the discretion of the court. See Slide 2 for further information on these forward-looking statements
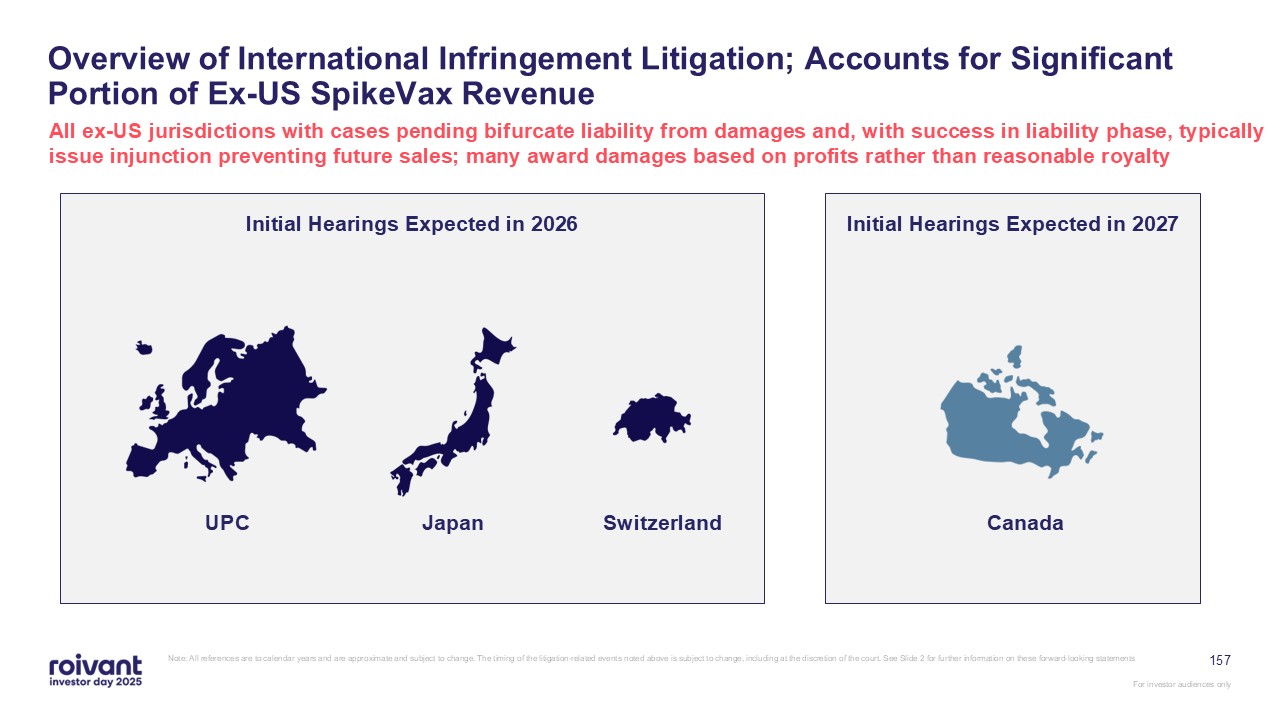
157 Overview of International Infringement Litigation; Accounts for Significant
Portion of Ex-US SpikeVax Revenue All ex-US jurisdictions with cases pending bifurcate liability from damages and, with success in liability phase, typically issue injunction preventing future sales; many award damages based on profits
rather than reasonable royalty Initial Hearings Expected in 2026 Initial Hearings Expected in 2027 UPC Japan Switzerland Canada Note: All references are to calendar years and are approximate and subject to change. The timing of the
litigation-related events noted above is subject to change, including at the discretion of the court. See Slide 2 for further information on these forward-looking statements

Pfizer Case
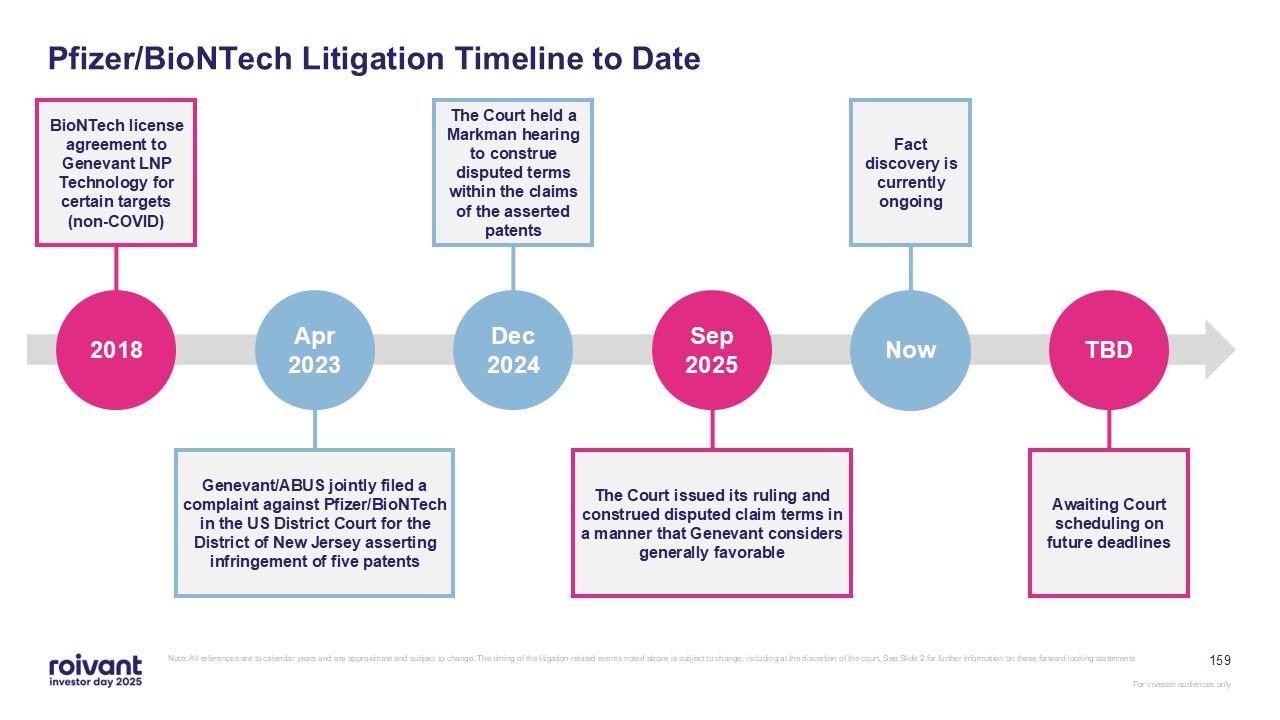
159 Pfizer/BioNTech Litigation Timeline to Date Note: All references are to
calendar years and are approximate and subject to change. The timing of the litigation-related events noted above is subject to change, including at the discretion of the court. See Slide 2 for further information on these forward-looking
statements Awaiting Court scheduling on future deadlines TBD The Court held a Markman hearing to construe disputed terms within the claims of the asserted patents Dec 2024 Now Fact discovery is currently ongoing BioNTech license
agreement to Genevant LNP Technology for certain targets (non-COVID) 2018 The Court issued its ruling and construed disputed claim terms in a manner that Genevant considers generally favorable Sep 2025 Genevant/ABUS jointly filed a
complaint against Pfizer/BioNTech in the US District Court for the District of New Jersey asserting infringement of five patents Apr 2023

Publicly Available Evidence Supports the Initial Filing of Our Case Publicly
Available Evidence Supporting the Case1 BioNTech had license to Genevant LNP Technology for use for specific cancer and rare liver disease targets dating back to 2018; contract described Genevant’s platform as “the best lipid nanoparticle
technology” FDA EUA filing indicated that Pfizer/BioNTech’s mRNA-vaccine infringes on Genevant and Arbutus lipid composition patents ‘651, ‘359 and ‘378 CNN report covering Pfizer’s manufacturing process confirmed infringement of Genevant
and Arbutus manufacturing patents ‘320 and ‘098 160 “T-Mixer” used to create LNP; Arbutus/Genevant ‘320 patent Note: Case 3:23-cv-01876-ZNQ-TJB, Document 1; COMPLAINT FOR PATENT INFRINGEMENT (2022)

161 In Summary: LNP Litigation We believe both Moderna’s and Pfizer/BioNTech’s
COVID-19 vaccines infringe multiple Genevant/Arbutus patents Trial/hearings in both the US and the ex-US litigation against Moderna expected in 2026 Awaiting court scheduling in the Pfizer/BioNTech litigation Markman decisions in both
cases viewed by Genevant generally to be favorable For investor audiences only Note: All references are to calendar years and are approximate and subject to change. The timing of the litigation-related events noted above is subject to
change, including at the discretion of the court. See Slide 2 for further information on these forward-looking statements

Richard Pulik CFO, Roivant 162 Financial Outlook
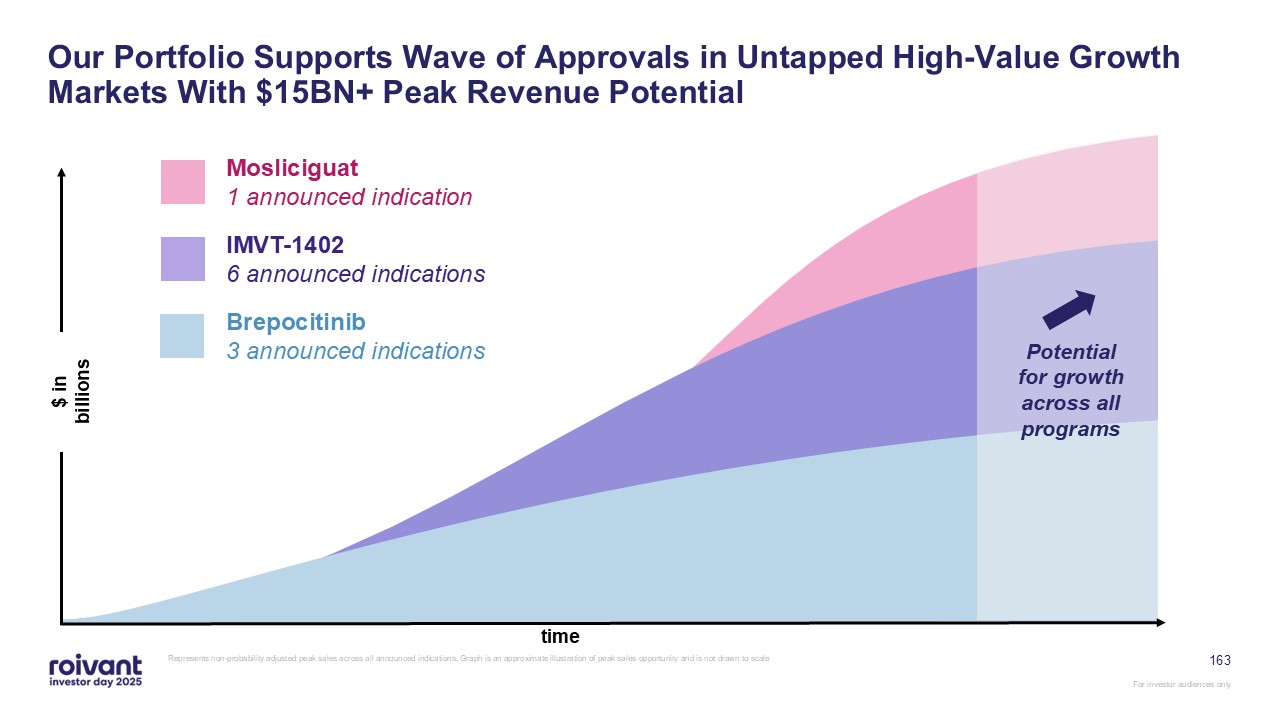
163 Our Portfolio Supports Wave of Approvals in Untapped High-Value Growth
Markets With $15BN+ Peak Revenue Potential Represents non-probability adjusted peak sales across all announced indications. Graph is an approximate illustration of peak sales opportunity and is not drawn to scale time $ in
billions Potential for growth across all programs Mosliciguat 1 announced indication IMVT-1402 6 announced indications Brepocitinib 3 announced indications

164 Roivant Capitalized to Profitability With $4.4BN Cash Balance to Advance
Current Priorities and Fund Selective Capital Allocation Opportunities1 1. Cash, cash equivalents, restricted cash, and marketable securities as of September 30, 2025 Invest in Current Pipeline & Launches 10+ disclosed indications in
mid-late-stage development Invest in New Opportunities ~$2BN available for late-stage development and high value creation opportunities Return Excess Capital to Shareholders $1.5BN buyback completed and additional $500M
authorized + + Roivant-Led Immunovant Financing Generated Gross Proceeds to Immunovant of Approximately $550M, Extending Immunovant’s Cash Runway to the Launch of IMVT-1402 in Graves’ Disease
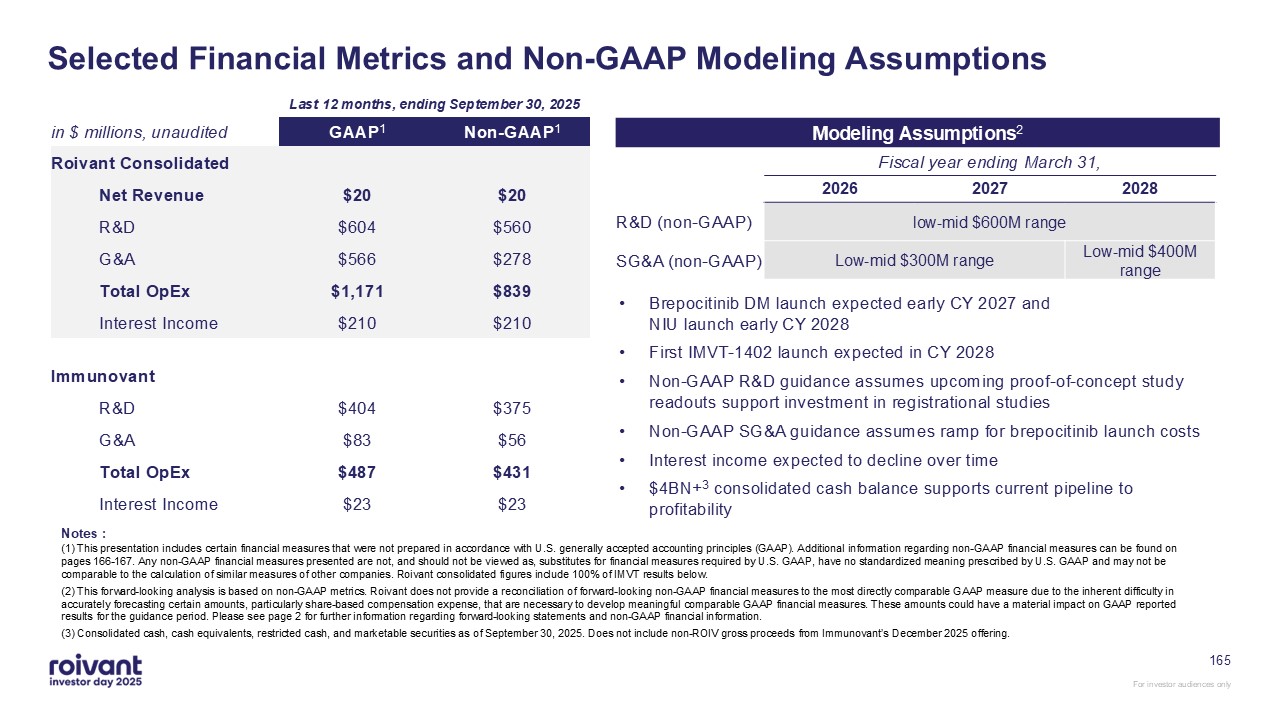
Modeling Assumptions2 in $ millions, unaudited GAAP1 Non-GAAP1 Roivant
Consolidated Net Revenue $20 $20 R&D $604 $560 G&A $566 $278 Total OpEx $1,171 $839 Interest Income $210 $210 Immunovant R&D $404 $375 G&A $83 $56 Total OpEx $487 $431 Interest
Income $23 $23 165 Selected Financial Metrics and Non-GAAP Modeling Assumptions Last 12 months, ending September 30, 2025 2026 2027 2028 low-mid $600M range Low-mid $300M range Low-mid $400M range Fiscal year ending March
31, Brepocitinib DM launch expected early CY 2027 and NIU launch early CY 2028 First IMVT-1402 launch expected in CY 2028 Non-GAAP R&D guidance assumes upcoming proof-of-concept study readouts support investment in registrational
studies Non-GAAP SG&A guidance assumes ramp for brepocitinib launch costs Interest income expected to decline over time $4BN+3 consolidated cash balance supports current pipeline to profitability R&D (non-GAAP) SG&A
(non-GAAP) (1) This presentation includes certain financial measures that were not prepared in accordance with U.S. generally accepted accounting principles (GAAP). Additional information regarding non-GAAP financial measures can be found on
pages 166-167. Any non-GAAP financial measures presented are not, and should not be viewed as, substitutes for financial measures required by U.S. GAAP, have no standardized meaning prescribed by U.S. GAAP and may not be comparable to the
calculation of similar measures of other companies. Roivant consolidated figures include 100% of IMVT results below. (2) This forward-looking analysis is based on non-GAAP metrics. Roivant does not provide a reconciliation of
forward-looking non-GAAP financial measures to the most directly comparable GAAP measure due to the inherent difficulty in accurately forecasting certain amounts, particularly share-based compensation expense, that are necessary to develop
meaningful comparable GAAP financial measures. These amounts could have a material impact on GAAP reported results for the guidance period. Please see page 2 for further information regarding forward-looking statements and non-GAAP financial
information. (3) Consolidated cash, cash equivalents, restricted cash, and marketable securities as of September 30, 2025. Does not include non-ROIV gross proceeds from Immunovant’s December 2025 offering. Notes :
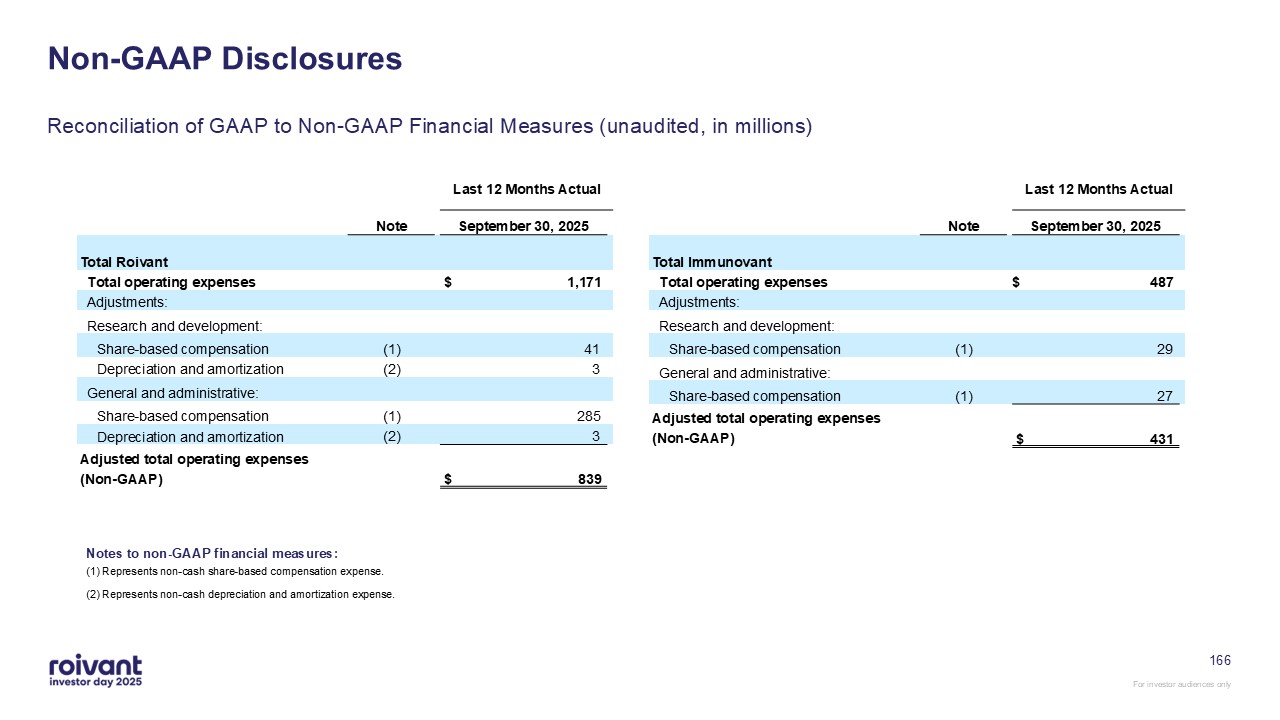
Non-GAAP Disclosures (1) Represents non-cash share-based compensation
expense. (2) Represents non-cash depreciation and amortization expense. Notes to non-GAAP financial measures: Reconciliation of GAAP to Non-GAAP Financial Measures (unaudited, in millions) 166 Last 12 Months
Actual Note September 30, 2025 Total Roivant Total operating expenses $ 1,171 Adjustments: Research and development: Share-based compensation (1) 41 Depreciation and
amortization (2) 3 General and administrative: Share-based compensation (1) 285 Depreciation and amortization (2) 3 Adjusted total operating expenses (Non-GAAP) $ 839 Last 12 Months
Actual Note September 30, 2025 Total Immunovant Total operating expenses $ 487 Adjustments: Research and development: Share-based compensation (1) 29 General and
administrative: Share-based compensation (1) 27 Adjusted total operating expenses (Non-GAAP) $ 431

167 Non-GAAP Disclosures Reconciliation of GAAP to Non-GAAP Financial Measures
(unaudited, in millions) Last 12 Months Actual Note September 30, 2025 Total Roivant Research and development expenses $ 604 Adjustments Share-based compensation (1) 41 Depreciation and
amortization (2) 3 Adjusted research and development expenses (Non-GAAP) $ 560 Last 12 Months Actual Note September 30, 2025 Immunovant Research and development expenses $ 404
Adjustments Share-based compensation (1) 29 Adjusted research and development expenses (Non-GAAP) $ 375 Last 12 Months Actual Note September 30, 2025 Total Roivant General and
administrative expenses $ 566 Adjustments Share-based compensation (1) 285 Depreciation and amortization (2) 3 Adjusted research and development expenses (Non-GAAP) $ 278 Last 12 Months
Actual Note September 30, 2025 Immunovant General and administrative expenses $ 83 Adjustments Share-based compensation (1) 27 Adjusted research and development expenses (Non-GAAP) $ 56 (1)
Represents non-cash share-based compensation expense. (2) Represents non-cash depreciation and amortization expense. Notes to non-GAAP financial measures:

Matt Gline CEO, Roivant 168 Closing Remarks
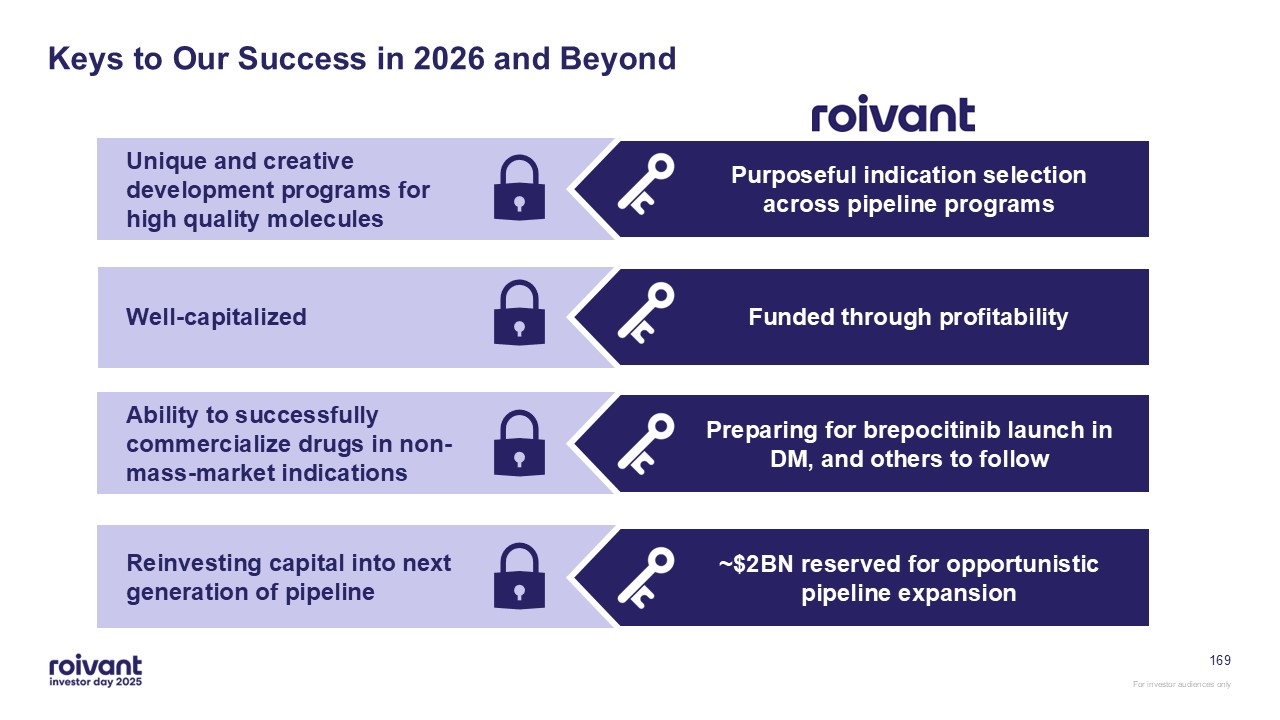
169 Keys to Our Success in 2026 and Beyond Well-capitalized Funded through
profitability Unique and creative development programs for high quality molecules Purposeful indication selection across pipeline programs Ability to successfully commercialize drugs in non-mass-market indications Preparing for
brepocitinib launch in DM, and others to follow Reinvesting capital into next generation of pipeline ~$2BN reserved for opportunistic pipeline expansion
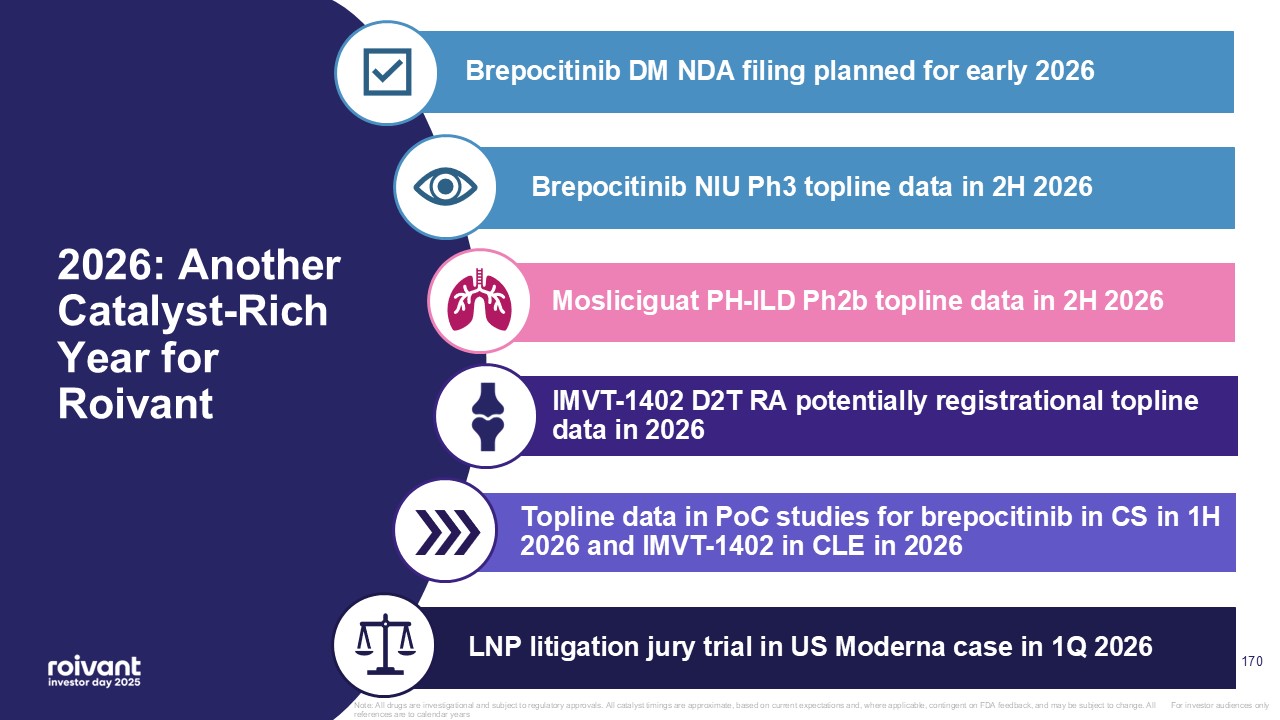
2026: Another Catalyst-Rich Year for Roivant For investor audiences only LNP
litigation jury trial in US Moderna case in 1Q 2026 Mosliciguat PH-ILD Ph2b topline data in 2H 2026 Brepocitinib NIU Ph3 topline data in 2H 2026 Brepocitinib DM NDA filing planned for early 2026 Topline data in PoC studies for
brepocitinib in CS in 1H 2026 and IMVT-1402 in CLE in 2026 IMVT-1402 D2T RA potentially registrational topline data in 2026 Note: All drugs are investigational and subject to regulatory approvals. All catalyst timings are approximate, based
on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar years 170
171 Roivant’s Priorities for the Next 12 Months… Prepare for brepocitinib
launch Progress IMVT-1402 development across multiple pivotal studies Convert multiple PoC studies across 3 products into pivotal programs Execute on LNP litigation Add to existing pipeline For investor audiences only
172 … To Fully Capitalize on an Exciting Next Decade Represents
non-probability adjusted peak sales across all announced indications. Graph is an approximate illustration of peak sales opportunity and is not drawn to scale time $ in billions Potential for growth across all programs Mosliciguat 1
announced indication IMVT-1402 6 announced indications Brepocitinib 3 announced indications
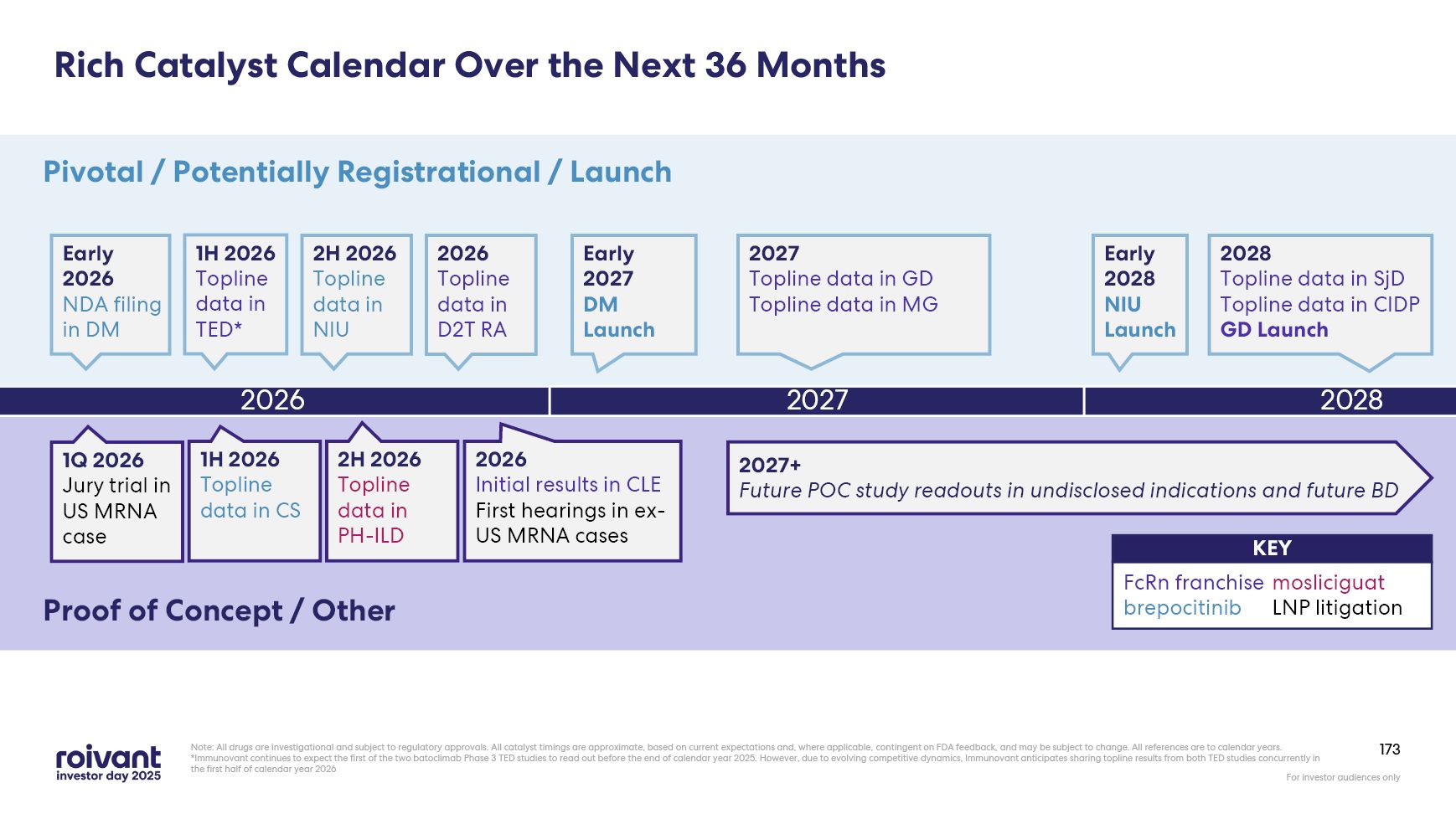
Rich Catalyst Calendar Over the Next 36 Months Note: All drugs are
investigational and subject to regulatory approvals. All catalyst timings are approximate, based on current expectations and, where applicable, contingent on FDA feedback, and may be subject to change. All references are to calendar
years.*Immunovant continues to expect the first of the two batoclimab Phase 3 TED studies to read out before the end of calendar year 2025. However, due to evolving competitive dynamics, Immunovant anticipates sharing topline results from
both TED studies concurrently in the first half of calendar year 2026 2026 Pivotal / Potentially Registrational / Launch Proof of Concept / Other 1H 2026 Topline data in TED* 2027 2028 2H 2026 Topline data in PH-ILD 1Q 2026 Jury
trial in US MRNA case 2026 Topline data in D2T RA 2026 Initial results in CLE First hearings in ex-US MRNA cases 2H 2026 Topline data in NIU 2027 Topline data in GD Topline data in MG 2028 Topline data in SjD Topline data in
CIDP GD Launch FcRn franchise brepocitinib mosliciguat LNP litigation KEY 1H 2026 Topline data in CS Early 2028 NIU Launch Early 2027 DM Launch 2027+ Future POC study readouts in undisclosed indications and future BD
173 Early 2026 NDA filing in DM
Q&A

Appendix

Selected Study Designs

177 Positive topline results announced in September 2025 Note: Additional
inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide DM: dermatomyositis; QD: daily; OCS: oral corticosteroids; CDASI: Cutaneous Dermatomyositis Disease Area and
Severity Index; DMOMS: Dermatomyositis Outcomes for Muscle and Skin N = 241 Adults with active DM with both muscle and skin disease 1:1:1 Randomization Placebo Brepocitinib 30 mg QD Brepocitinib 15 mg QD : Single Phase 3 Study for
Brepocitinib in Dermatomyositis Primary Endpoint: Mean Total Improvement Score (TIS) at Week 52 Secondary Endpoints: CDASI Activity Score DMOMS TIS ≥ 40 TIS ≥ 60 52 Week 0 Steroid taper: Mandatory OCS taper to ≤5 mg/day from
week 12 to 36; recommended further tapering at investigator discretion

178 : : Phase 3 Study for Brepocitinib in Non-Infectious Uveitis Two
identical sub-studies, CLARITY-1 and CLARITY-2, under a single protocol Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide QD: daily; OCS: oral
corticosteroids; CST: central subfield thickness; WFFA: wide-field fluorescein angiography References are to calendar years N = 371 CLARITY-1 N = 180 CLARITY-2 N = 191 Adults with active non-infectious intermediate-, posterior-, or
panuveitis Placebo Brepocitinib 45 mg QD Primary Endpoint: Time to Treatment Failure Secondary Endpoints: Include measurements related to Macular edema/CST Retinal vascular leakage as measured through WFFA Functional vision 1:1
Randomization 48 Week 0 Steroid burst and taper: 60 mg/day OCS burst for 14 days; forced taper to 0 mg/day by Week 8 (identical to Phase 2 study)

179 BEACON: Phase 2 Study for Brepocitinib in Cutaneous Sarcoidosis Note:
Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide CS: cutaneous sarcoidosis; QD: daily; CFB: change from baseline; CSAMI: Cutaneous Sarcoidosis
Activity and Morphology Instrument References are to calendar years N = 31 Adults with CS Placebo Brepocitinib 45 mg QD Brepocitinib 15 mg QD Primary Efficacy Endpoint: CFB in CSAMI Score at Week 16 for 45 mg brepocitinib 3:2:2
Randomization 16 Week 0
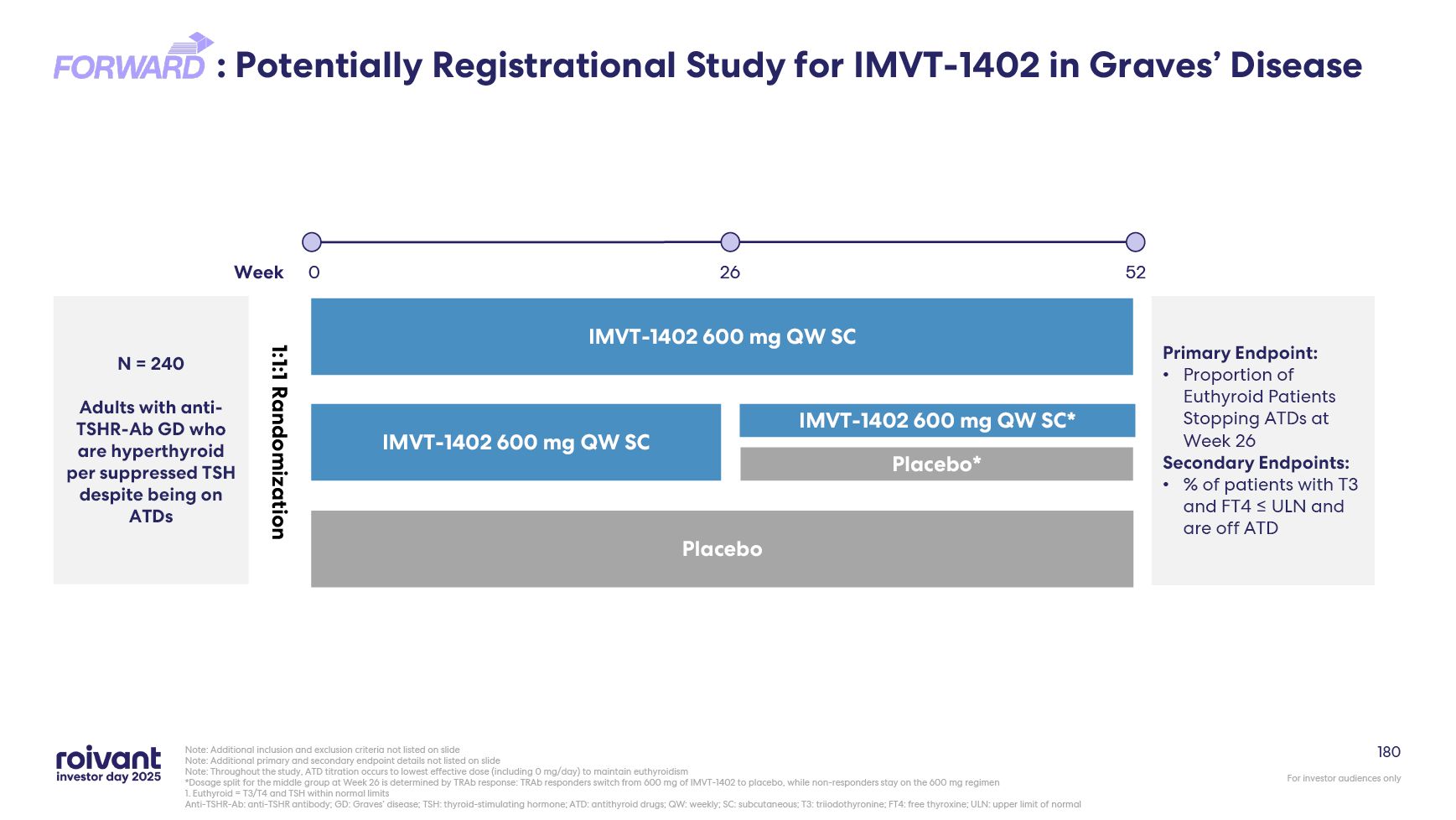
180 : Potentially Registrational Study for IMVT-1402 in Graves’ Disease
Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide Note: Throughout the study, ATD titration occurs to lowest effective dose (including 0
mg/day) to maintain euthyroidism *Dosage split for the middle group at Week 26 is determined by TRAb response: TRAb responders switch from 600 mg of IMVT-1402 to placebo, while non-responders stay on the 600 mg regimen 1. Euthyroid = T3/T4
and TSH within normal limits Anti-TSHR-Ab: anti-TSHR antibody; GD: Graves’ disease; TSH: thyroid-stimulating hormone; ATD: antithyroid drugs; QW: weekly; SC: subcutaneous; T3: triiodothyronine; FT4: free thyroxine; ULN: upper limit of
normal N = 240 Adults with anti-TSHR-Ab GD who are hyperthyroid per suppressed TSH despite being on ATDs 52 26 IMVT-1402 600 mg QW SC Placebo IMVT-1402 600 mg QW SC IMVT-1402 600 mg QW SC* Placebo* Week Primary Endpoint:
Proportion of Euthyroid Patients Stopping ATDs at Week 26 Secondary Endpoints: % of patients with T3 and FT4 ≤ ULN and are off ATD 1:1:1 Randomization 0

181 II: Potentially Registrational Study for IMVT-1402 in Graves’
Disease Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide Note: Throughout the study, ATD titration occurs to lowest effective dose (including
0 mg/day) to maintain euthyroidism 1. Euthyroid = T3/T4 and TSH within normal limits Anti-TSHR-Ab: anti-TSHR antibody; GD: Graves’ disease; TSH: thyroid-stimulating hormone; ATD: antithyroid drugs; QW: weekly; SC: subcutaneous; T3:
triiodothyronine; FT4: free thyroxine; ULN: upper limit of normal N = 210 Adults with anti-TSHR-Ab GD who are hyperthyroid per suppressed TSH despite being on ATDs Placebo IMVT-1402 600 mg QW SC IMVT-1402 300 mg QW
SC 26 Week 0 Primary Endpoint: Proportion of 600 mg Patients who are Euthyroid and Stopping ATDs at Week 26 Secondary Endpoints: % of patients with T3 and FT4 ≤ ULN and are off ATD 1:1:1 Randomization
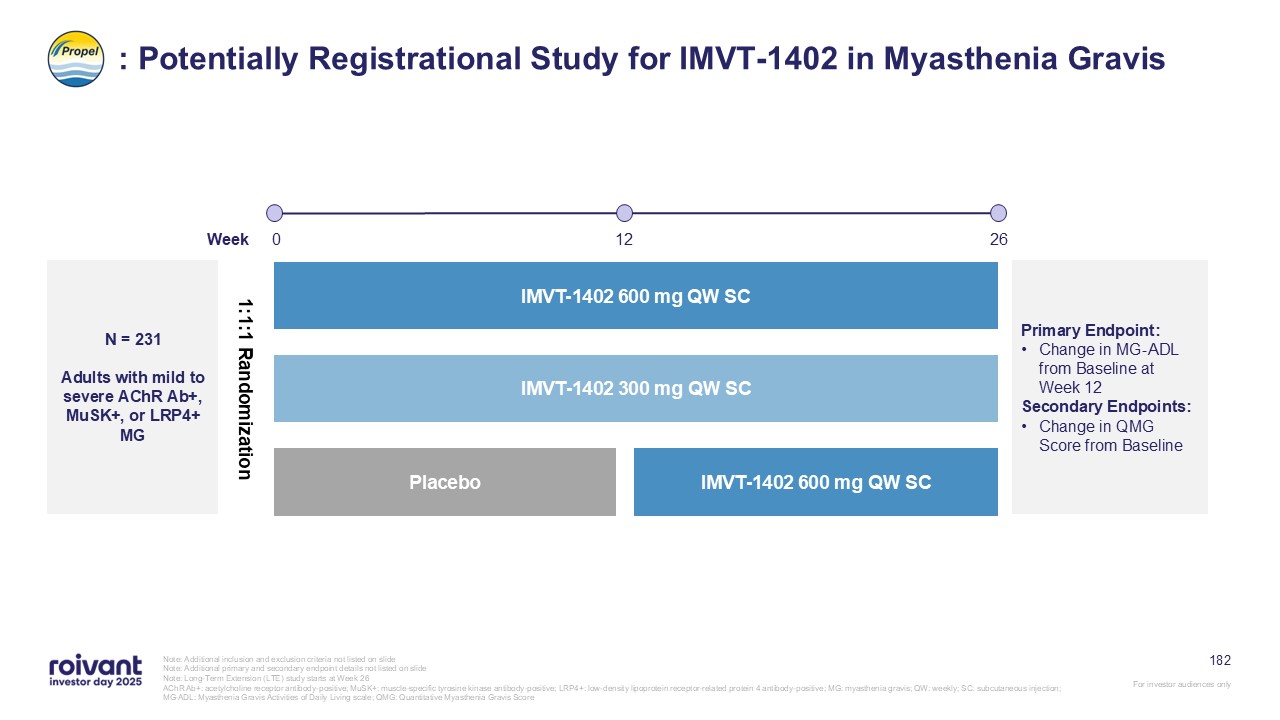
182 : Potentially Registrational Study for IMVT-1402 in Myasthenia Gravis
Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide Note: Long-Term Extension (LTE) study starts at Week 26 AChR Ab+: acetylcholine receptor
antibody-positive; MuSK+: muscle-specific tyrosine kinase antibody-positive; LRP4+: low-density lipoprotein receptor-related protein 4 antibody-positive; MG: myasthenia gravis; QW: weekly; SC: subcutaneous injection; MG-ADL: Myasthenia Gravis
Activities of Daily Living scale; QMG: Quantitative Myasthenia Gravis Score N = 231 Adults with mild to severe AChR Ab+, MuSK+, or LRP4+ MG 12 26 IMVT-1402 600 mg QW SC Placebo IMVT-1402 600 mg QW SC IMVT-1402 300 mg QW
SC Week 0 Primary Endpoint: Change in MG-ADL from Baseline at Week 12 Secondary Endpoints: Change in QMG Score from Baseline 1:1:1 Randomization
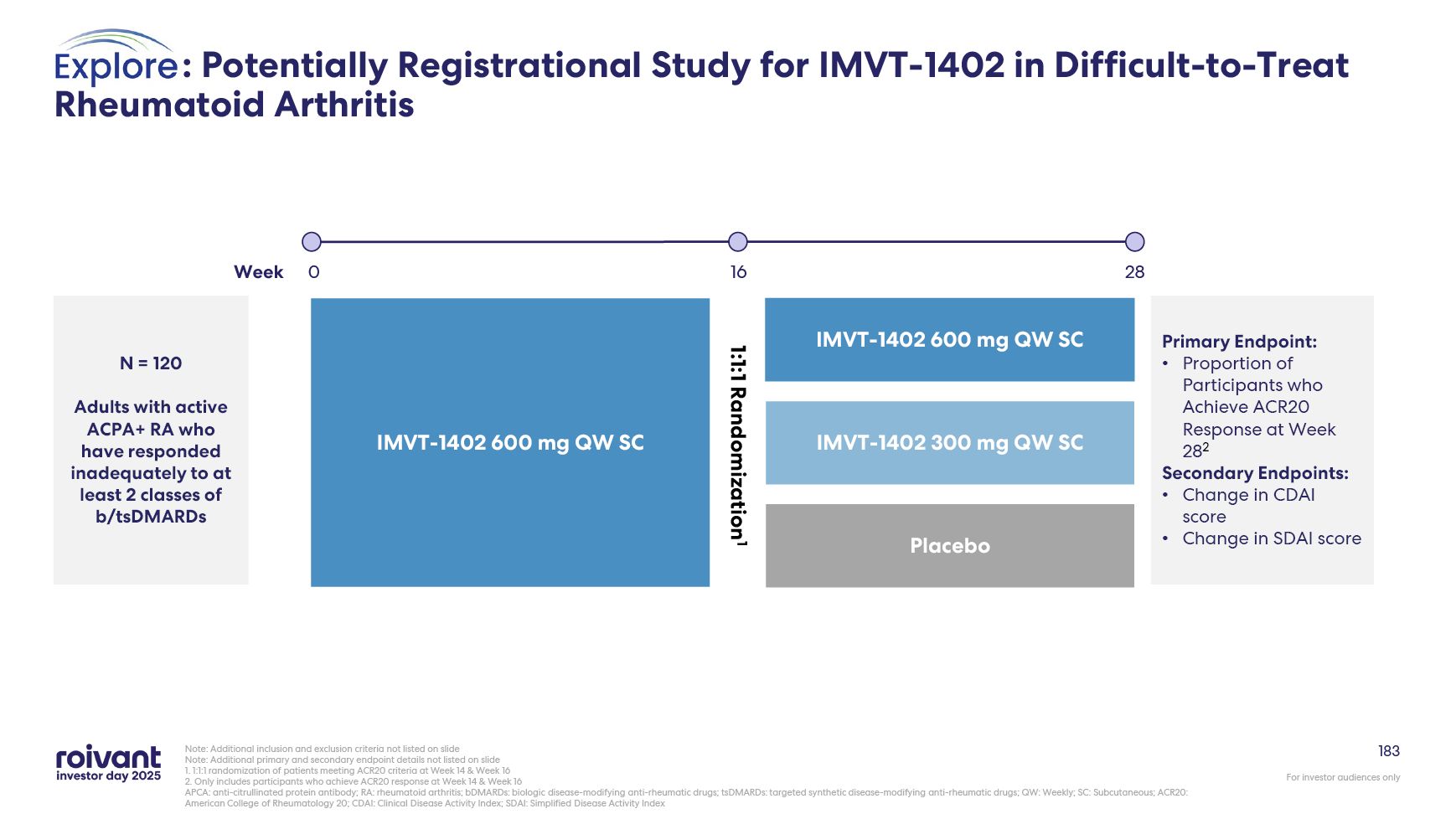
183 : Potentially Registrational Study for IMVT-1402 in Difficult-to-Treat
Rheumatoid Arthritis Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide 1. 1:1:1 randomization of patients meeting ACR20 criteria at Week 14
& Week 16 2. Only includes participants who achieve ACR20 response at Week 14 & Week 16 APCA: anti-citrullinated protein antibody; RA: rheumatoid arthritis; bDMARDs: biologic disease-modifying anti-rheumatic drugs; tsDMARDs:
targeted synthetic disease-modifying anti-rheumatic drugs; QW: Weekly; SC: Subcutaneous; ACR20: American College of Rheumatology 20; CDAI: Clinical Disease Activity Index; SDAI: Simplified Disease Activity Index N = 120 Adults with active
ACPA+ RA who have responded inadequately to at least 2 classes of b/tsDMARDs 1:1:1 Randomization1 16 28 IMVT-1402 600 mg QW SC IMVT-1402 300 mg QW SC Placebo IMVT-1402 600 mg QW SC Week 0 Primary Endpoint: Proportion of
Participants who Achieve ACR20 Response at Week 282 Secondary Endpoints: Change in CDAI score Change in SDAI score
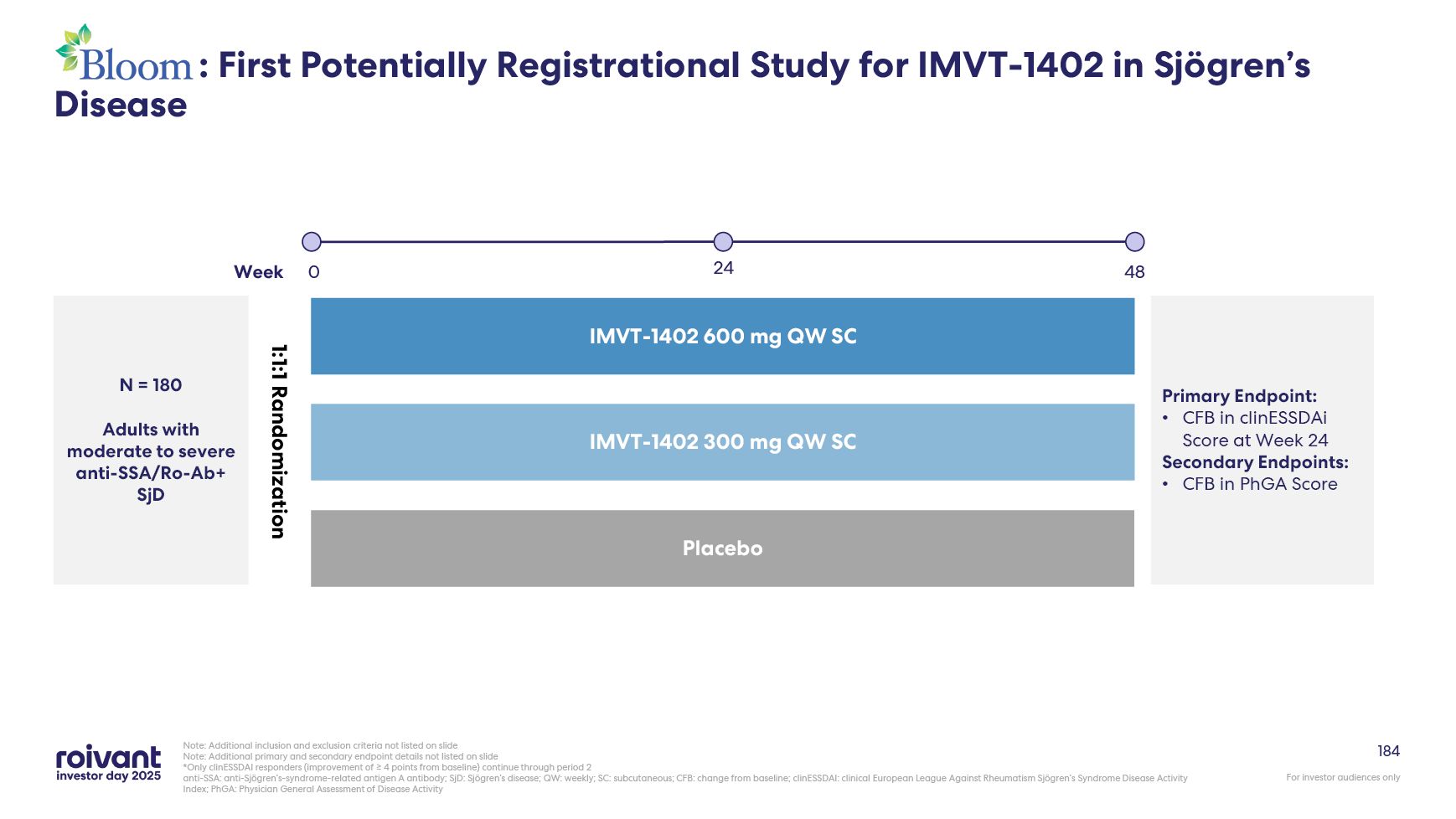
184 Note: Additional inclusion and exclusion criteria not listed on
slide Note: Additional primary and secondary endpoint details not listed on slide *Only clinESSDAI responders (improvement of ≥ 4 points from baseline) continue through period 2 anti-SSA: anti-Sjögren’s-syndrome-related antigen A antibody;
SjD: Sjögren’s disease; QW: weekly; SC: subcutaneous; CFB: change from baseline; clinESSDAI: clinical European League Against Rheumatism Sjögren’s Syndrome Disease Activity Index; PhGA: Physician General Assessment of Disease Activity
IMVT-1402 600 mg QW SC IMVT-1402 300 mg QW SC Placebo N = 180 Adults with moderate to severe anti-SSA/Ro-Ab+ SjD 48 Week 0 Primary Endpoint: CFB in clinESSDAi Score at Week 24 Secondary Endpoints: CFB in PhGA Score 1:1:1
Randomization : First Potentially Registrational Study for IMVT-1402 in Sjögren’s Disease 24

185 Note: Additional inclusion and exclusion criteria not listed on
slide Note: Additional primary and secondary endpoint details not listed on slide Note: Simplified study design without washout period and flare requirement prior to randomization based on experience in the batoclimab CIDP study in
identifying patients with active disease CIDP: chronic inflammatory demyelinating polyneuropathy; QW, once weekly; SC subcutaneous; aINCAT: Adjusted Inflammatory Neuropathy Cause and Treatment disability score; CFB: change from baseline;
I-RODS: Inflammatory Rash-Built Overall Disability Scale; MRC-SS: Medical Research Council Sum Score N = 162 Adults with active CIDP Placebo IMVT-1402 600 mg Qw SC 24 Week 0 Primary Endpoint: Proportion of Patients Remaining Relapse
Free (aINCAT) at Week 24 Secondary Endpoints: CFB in I-RODS CFB in Mean Grip Strength CFB in MRC-SS 2:1 Randomization : Potentially Registrational Study for IMVT-1402 in Chronic Inflammatory Demyelinating Polyneuropathy
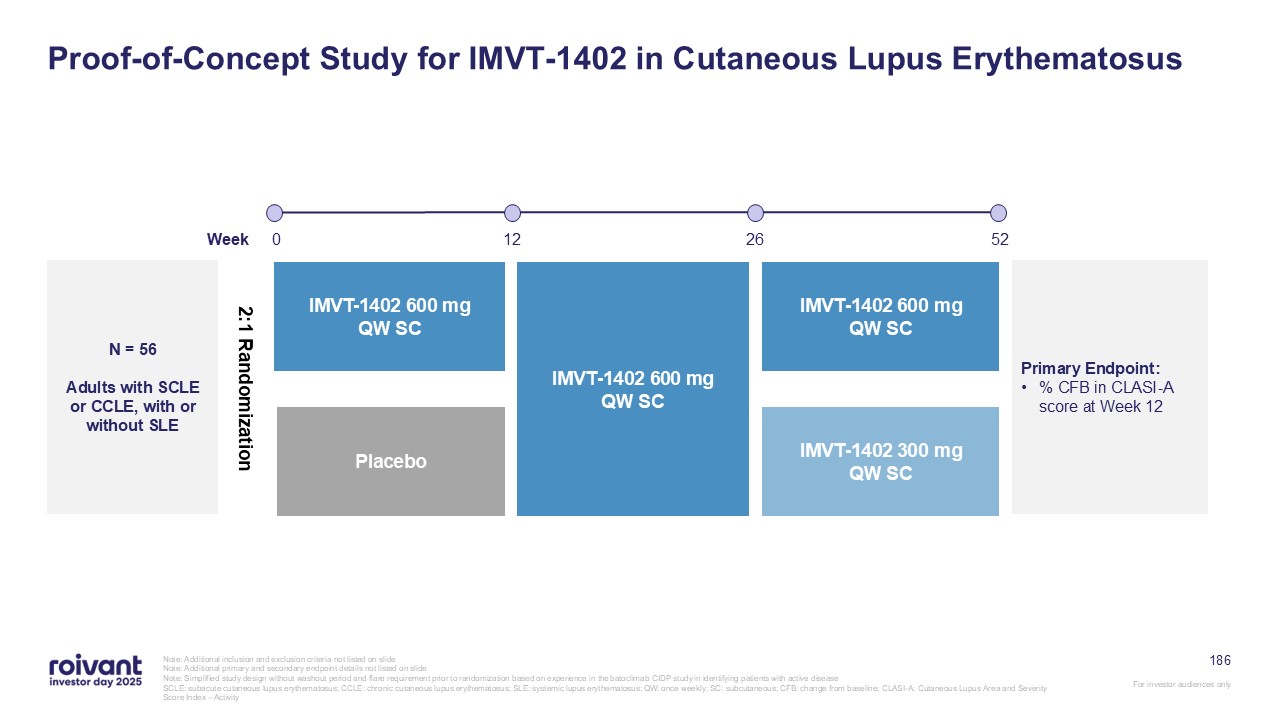
186 Proof-of-Concept Study for IMVT-1402 in Cutaneous Lupus
Erythematosus Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide Note: Simplified study design without washout period and flare requirement
prior to randomization based on experience in the batoclimab CIDP study in identifying patients with active disease SCLE: subacute cutaneous lupus erythematosus; CCLE: chronic cutaneous lupus erythematosus; SLE: systemic lupus erythematosus;
QW: once weekly; SC: subcutaneous; CFB: change from baseline; CLASI-A: Cutaneous Lupus Area and Severity Score Index – Activity N = 56 Adults with SCLE or CCLE, with or without SLE 52 12 26 Placebo IMVT-1402 600 mg Qw SC IMVT-1402 600
mg QW SC IMVT-1402 600 mg Qw SC IMVT-1402 300 mg Qw SC Week 0 Primary Endpoint: % CFB in CLASI-A score at Week 12 2:1 Randomization
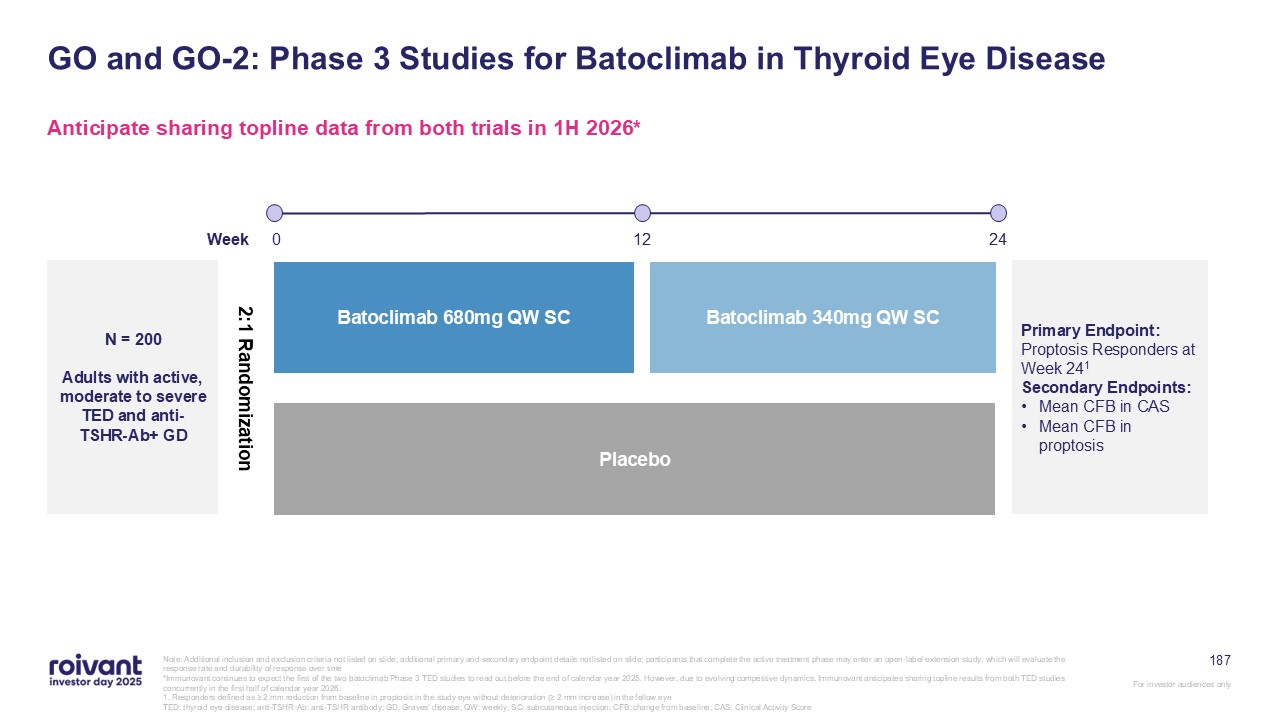
187 GO and GO-2: Phase 3 Studies for Batoclimab in Thyroid Eye
Disease Anticipate sharing topline data from both trials in 1H 2026* Note: Additional inclusion and exclusion criteria not listed on slide; additional primary and secondary endpoint details not listed on slide; participants that complete
the active treatment phase may enter an open-label extension study, which will evaluate the response rate and durability of response over time *Immunovant continues to expect the first of the two batoclimab Phase 3 TED studies to read out
before the end of calendar year 2025. However, due to evolving competitive dynamics, Immunovant anticipates sharing topline results from both TED studies concurrently in the first half of calendar year 2026. 1. Responders defined as ≥ 2 mm
reduction from baseline in proptosis in the study eye without deterioration (≥ 2 mm increase) in the fellow eye TED: thyroid eye disease; anti-TSHR-Ab: anti-TSHR antibody; GD: Graves’ disease; QW: weekly; SC: subcutaneous injection; CFB:
change from baseline; CAS: Clinical Activity Score N = 200 Adults with active, moderate to severe TED and anti-TSHR-Ab+ GD Batoclimab 680mg QW SC Batoclimab 340mg QW SC Placebo Primary Endpoint: Proptosis Responders at Week
241 Secondary Endpoints: Mean CFB in CAS Mean CFB in proptosis 2:1 Randomization 24 12 Week 0
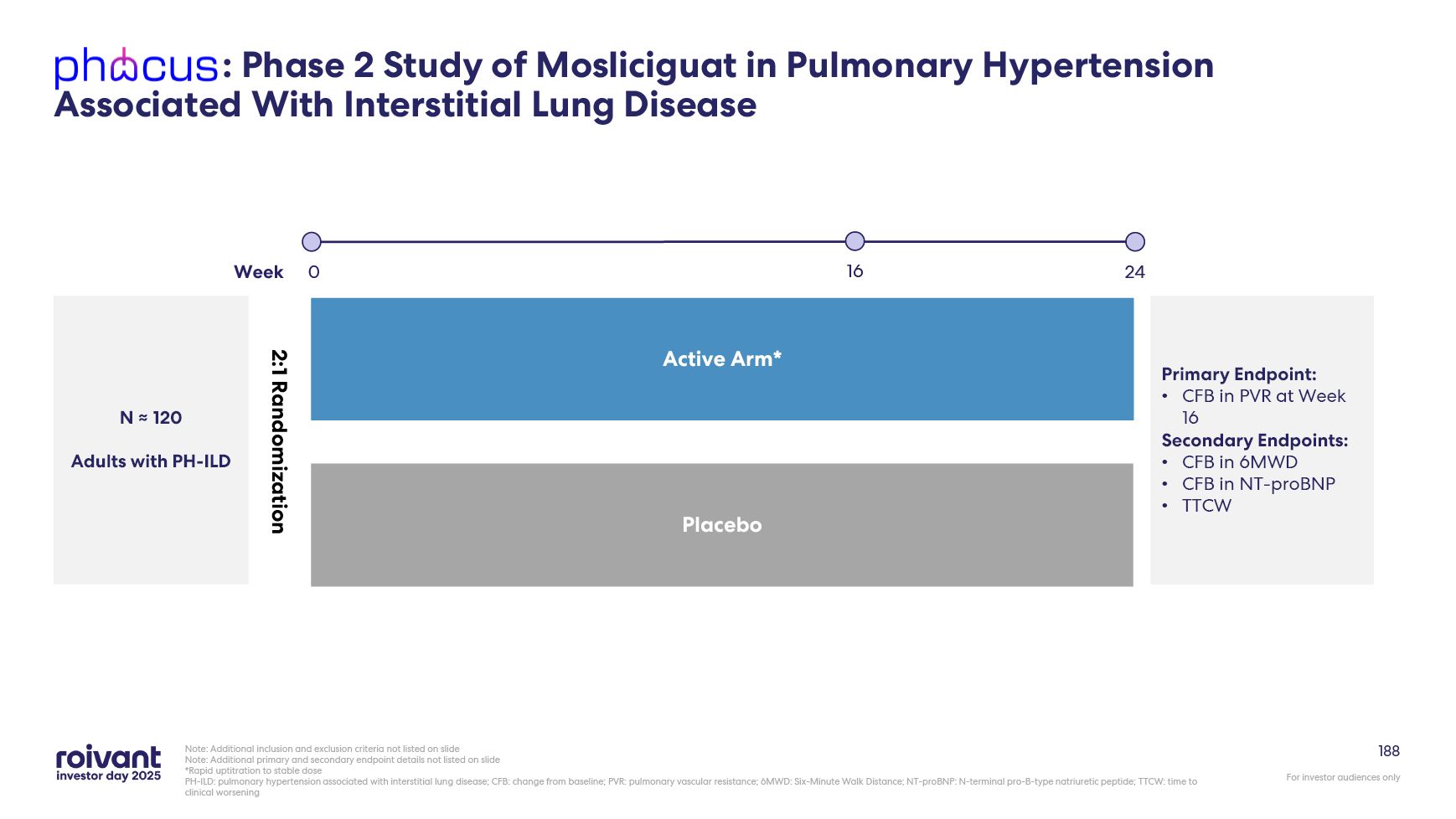
188 : Phase 2 Study of Mosliciguat in Pulmonary Hypertension Associated With
Interstitial Lung Disease Note: Additional inclusion and exclusion criteria not listed on slide Note: Additional primary and secondary endpoint details not listed on slide *Rapid uptitration to stable dose PH-ILD: pulmonary hypertension
associated with interstitial lung disease; CFB: change from baseline; PVR: pulmonary vascular resistance; 6MWD: Six-Minute Walk Distance; NT-proBNP: N-terminal pro-B-type natriuretic peptide; TTCW: time to clinical worsening N ≈ 120 Adults
with PH-ILD Active Arm* Placebo Week 24 0 Primary Endpoint: CFB in PVR at Week 16 Secondary Endpoints: CFB in 6MWD CFB in NT-proBNP TTCW 2:1 Randomization 16

Vant Financials Overview

190 Priovant: Other Details ROIV owns 73%1 of Priovant, with Pfizer owning
24%. Ownership Geographic Rights Intellectual Property Milestones Royalties 1. As of September 30, 2025. 65% on a fully diluted basis. 2. Includes potential patent term extension We expect brepocitinib to have US
exclusivity at least until 20392. Priovant has commercial rights to brepocitinib in US and Japan. Priovant is obligated to pay Pfizer mid tens-of-millions if sales exceed a mid hundreds-of-millions amount in Priovant territories. Pfizer
is obligated to pay Priovant low tens-of-millions if sales exceed a mid hundreds-of-millions amount in non-Priovant territories. Priovant is obligated to pay Pfizer tiered sub-teens royalties on annual sales in Priovant territories. Pfizer
is obligated to pay Priovant tiered high single digits to sub-teens royalties on annual sales in non-Priovant territories.

191 Immunovant: Other Details Immunovant is publicly traded, with ROIV owning
55%1 Ownership Geographic Rights Intellectual Property Milestones Royalties 1. As of September 30, 2025, 50% on a diluted basis 2. Immunovant has commercialization rights in U.S., Canada, Mexico, the E.U., the U.K.,
Switzerland, the Middle East, North Africa and Latin America 3. Not including any potential patent term extension We expect IMVT-1402 to have US exclusivity at least until 20433 Immunovant has global rights to batoclimab and IMVT-1402
outside of APAC2 Immunovant is obligated to pay HanAll future development and commercial milestone payments up to an aggregate $420M (of which $32.5M has been paid) Immunovant is obligated to pay HanAll tiered mid-single-digits to mid-teens
royalty on net sales of batoclimab and IMVT-1402

192 Pulmovant: Other Details ROIV owns 98%1 of Pulmovant. Ownership
Geographic Rights Intellectual Property Milestones Royalties 1. As of September 30, 2025. 91% on a fully diluted basis. 2. Includes potential patent term extension We expect mosliciguat to have US exclusivity until the
mid-2040s2. Pulmovant holds worldwide commercial rights to mosliciguat. Pulmovant is obligated to pay Bayer development, regulatory and net sales milestones, up to an aggregate $280M Pulmovant is obligated to pay Bayer tiered
high-single-digit royalties on annual net sales