SECURITIES AND EXCHANGE COMMISSION
FORM 10-K
(Mark One)
| [X] | ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2019
or
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
(Commission file number): 001-33635
![]()
GENE BIOTHERAPEUTICS INC.
(Exact name of registrant as specified in its charter)
(State of incorporation) |
(IRS Employer Identification No.) | |
San Diego, California 92121 |
||
(Address of principal executive offices) |
||
Securities
registered under Section
Securities
registered under Section
Common Stock, par value $0.0001 per share
Indicate
by check mark if the
Indicate
by check mark if the
Indicate
by check mark whether the
Indicate
by checkmark whether the Indicate
by check mark whether the Non-accelerated
filer
Emerging
Growth Company
[ ]
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for
complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate
by check mark whether the Registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report. [ ] Indicate
by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). [ ] Yes [X]
No The
aggregate market value of As
of March
TABLE
OF CONTENTS We
are filing this comprehensive Annual Report on Form 10-K for the fiscal year ended December 31, 2019 with expanded financial and
other disclosures in lieu of filing a separate Annual Report on Form 10-K for the fiscal years ended December 31, 2018 and December
31, 2017, and separate Quarterly Reports on Form 10-Q for the quarterly periods ended March 31, June 30, and September 30 during
2019, 2018 and 2017. Unless context requires otherwise, all references in this report to the
Due to financial hardship, we were unable to secure The
filing of this report will not result in us becoming “current” in our reporting requirements under the Securities
Exchange Act of 1934. It is our intention to become current, This
report, including the sections entitled “Business”, “Risk Factors”, and “Management’s Discussion
and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements our
ability to fund operations and business plans, and the timing of any funding or corporate development transactions we may
pursue; the
protection expected from our intellectual property rights and those of others, including actual or potential competitors;
and Caution
should be taken not to place undue reliance on any such forward-looking statements. Forward-looking statements are subject to
certain events, risks, and uncertainties that may be outside of our control that could cause actual results to differ materially
from those expressed in or implied by the forward-looking statements. These factors include, among others, the risks described
under Item 1A and elsewhere in this report, as well as in other reports and documents we file with the United States Securities
and Exchange Commission (the “SEC -2-
●
planned
development pathways and potential commercialization activities or opportunities for our product candidates;
●
the
timing, conduct and outcome of
●
the
anticipated results of our clinical studies and trials, as well as our expectations concerning the safety and efficacy of
our products and product candidates;
●
our
ability to generate revenues, and raise sufficient financing, maintain stock price and valuation, and to regain the listing
of our common stock on a national exchange;
●
our
ability to enter into acceptable relationships with one or more contract manufacturers
●
our
ability to enter into acceptable relationships with one or more development or commercialization partners to advance the commercialization
of new products and product candidates and the timing of any product launches;
●
our
growth, expansion and acquisition strategies, the success of such strategies, and the benefits we believe can be derived from
such strategies;
●
statements
that are not statements of historical facts. -3-
Our
lead product candidate Generx [Ad5FGF-4] is an angiogenic gene therapy product candidate designed for medical revascularization
for the potential treatment of patients with myocardial ischemia and refractory angina due to advanced coronary artery disease History We
were incorporated in Delaware in 2003. In 2006, we changed our name to Cardium Therapeutics Inc. In 2013, we changed the Company’s
name to Taxus Cardium Pharmaceuticals Group Inc. to reflect a broadened business plan During
the period covered by this report, our operations have been conducted principally through operating subsidiaries including the
following: We
entered 2017 in a cash constrained position. At that time, our principal operating goal was to secure the capital necessary to
advance the clinical development and commercialization of Generx. In October 2017 we entered into an agreement with Landmark Pegasus,
Inc. (“Landmark”), a business development and strategic partnering company, to assist us with the previously announced
plans to sell our Excellagen product and assist with the strategic partnering for the development of Generx. In lieu of a cash
engagement fee, we transferred our residual investment in LifeAgain along with our minority equity investment in Healthy Brands
to Landmark, effectively exiting those businesses. In
July 2018, we sold our FDA-cleared Excellagen® product to Olaregen Therapeutix, Inc. (“Olaregen”) for aggregate
consideration of up to $4,000,000. At closing, we received a cash payment of $650,000, the remaining to be paid as royalty payments
of 10% of all worldwide sales of Excellagen totaling up to an additional $3,350,000. As of the date of this report, no royalties
have been received. We retained rights to manufacture, market and sell Excellagen in Greater China, The Russian Federation, and
the Commonwealth of Independent States (Armenia, Azerbaijan, Belarus, Kazakhstan, Kyrgyzstan, Moldova, Tajikistan, Turkmenistan,
and Uzbekistan).
In
April 2020, after the period covered by this report, we transferred our residual rights in Excellagen to Shanxi Taxus Pharmaceuticals
Co. Ltd. (“Shanxi”) in exchange for the release of any rights or claims in ownership interest in Gene Biotherapeutics.
In connection with this transaction, Shanxi agreed to apply its previously funded $600,000 subscription payment as cash consideration
in exchange for the Excellagen ownership rights. Shanxi also released any future rights or claims against us. On
April 10, 2020, after the period covered by this report, our Angionetics, Inc. subsidiary entered into a Distribution and License
Agreement with Shanxi (as amended, the “Shanxi License Agreement”), granting Shanxi certain license rights with respect
to our Generx product candidate. The distribution and license rights commence only after we obtain U.S. FDA approval for marketing
and sale of Generx in the United States. The license rights include (a) a non-exclusive right to manufacture Generx products in
China, and (b) an exclusive right to market and sell Generx products in Singapore, Macau, Hong Kong, Taiwan, any other municipality
other than mainland China where Chinese (Mandarin or Cantonese) is the common language, the Russian Federation, and the Commonwealth
of Independent States (the “CIS”). The Shanxi License Agreement provides for a progress royalty ranging from
5% up to 10% based on annual net sales up to and including $50 million at 5%; 6% for sales ranging greater than $50 million to
$200 million; 8% for sales greater than $200 million to $450 million and at 10% for any sales greater than $450 million of the
Generx product sold by Shanxi in the licensed territory. In
May 2020, after the period covered by this report, we entered into a Preferred Stock Purchase Agreement with Nostrum Pharmaceuticals,
LLC (“Nostrum”), selling Nostrum 1,700,000 shares of our newly authorized Series B Convertible Preferred Stock in
exchange for $1,700,000. Each share of Series B Convertible Preferred Stock is convertible into shares of Common Stock at a conversion
ratio of 0.0113. Consequently the 1,700,000 shares are convertible into an aggregate of 150,442,478 shares of Common Stock. In
addition, Nostrum entered into an agreement with Sabby Healthcare Master Fund Ltd. (“Sabby”), the sole holder of our
outstanding Series A Convertible Preferred Stock, under which Nostrum purchased 220 shares of our Series A Convertible Preferred
Stock from Sabby, which is convertible into 88,496 shares of common stock. Consequently, the 220 shares are convertible into an
aggregate of 19,469,026 shares of Common Stock. Nostrum also agreed to purchase up to 570 additional Series A Convertible Preferred
Stock from Sabby, within one year following the effective date of the transaction. Since May 2020, 397 shares of Series
A Preferred Stock have been converted into 35,132,755 shares of our Common Stock (conversion rate of 88,496), that has
increased our outstanding Common Stock to 49,622,154 shares as of March 31, 2021. As a result of these
transactions, Nostrum currently controls approximately 75.2% of the voting interests of our Company. Nostrum
is a privately held pharmaceutical company engaged in the formulation and commercialization of specialty pharmaceutical products
and controlled release, orally administered, branded and generic drug products. We will use the proceeds from the sale of the
Series B Convertible Preferred Stock to fund working capital requirements in preparation for conducting the U.S. FDA-approved
Phase 3 clinical trial for our Generx product candidate, and a portion of these proceeds will be used to complete the financial
statements and disclosures in this report. We believe that Nostrum’s assets and experience in the formulation and commercialization
of pharmaceutical products will facilitate the administration and completion of the Phase 3 clinical trial for Generx on a cost-effective
basis. In
March 2021, after the period covered by this report, the Company entered into an agreement with FUJIFILM Diosynth Biotechnologies
(“FDB”) to manufacture the Generx [Ad5FGF-4] angiogenic gene therapy product candidate for Phase 3 clinical evaluation
for the treatment of refractory angina due to late-stage coronary artery disease. Manufacturing operations will be conducted at
FDB’s facilities in College Station, Texas where FDB will perform technology transfer and process development activities
for Phase 3 clinical and commercial-scale GMP manufacturing of Generx. Accordingly,
our current business is centered around the clinical development and commercialization of Generx for the potential treatment of
patients with myocardial ischemia and refractory angina due to advanced, late-stage coronary artery disease. In the future, we
expect to pursue other potential ischemia-related cardiovascular and cerebral therapeutic opportunities as well as advanced tissue
engineering applications. We estimate that there are up to 1.2 million patients in the U.S. with refractory angina, representing
up to $6.0 billion addressable market opportunity, and up to $20.0 billion worldwide. The
Generx Product Candidate Our
lead product candidate, Generx, is a first in class, single dose, angiogenic gene therapy product candidate that is designed to
improve blood flow and to increase the
Medical
Revascularization for Refractory Angina
The
Ad5FGF-4 product candidate The
transfected heart cells then express and release FGF-4 protein, which we believe promotes the growth of new blood vessels and
increased blood flow to ischemic heart tissue. The evidence shows that FGF-4 expressed by Ad5FGF-4 has the capacity to enlarge
pre-existing collateral arterioles (arteriogenesis) and to form new capillary vessels (angiogenesis) when driven by cardiac hemodynamic-impairment
and ischemic stimuli. In a pig model of myocardial ischemia, adenovirus mediated FGF gene therapy promoted increased regional
myocardial blood flow, as measured by contrast echocardiography, that correlated with an increase in capillary number, determined
by histologic assessment. Stimulation of angiogenesis by Ad5FGF-4 has also been demonstrated in an in vitro assay that recapitulates
all phases of the in vivo angiogenesis process and provides a functional bioassay for Ad5FGF-4. This assay demonstrates a synergistic
interaction between FGF-4 expressed by Ad5FGF-4, and endogenous vascular endothelial growth factor (VEGF) in the promotion of
neo-vessel formation, with evidence that FGF-4 controls angiogenesis upstream of VEGF. FGF-4 appears to be a key angiogenic regulatory
protein that stimulates the release and action of other angiogenic factors, including vascular endothelial growth factors (VEGF),
platelet-derived growth factors (PDGF), and hepatocyte growth factor (HGF), to orchestrate and promote the growth of a functional
collateral network in ischemic cardiac tissue. Generx
is administered to patients during a simple one-hour angiogram-like procedure by an interventional Addressable
Market Generx
is expected to initially target patients with refractory angina—chronic and disabling angina that: (1) are no longer responsive
to small molecule anti-anginal drug therapy, Proposed
Generx Treatment Algorithm for Patients with Refractory Angina Consistent
with Positioning in FDA-Cleared U.S. Phase 3 Clinical Trial Given
the widespread use of lipid-lowering drugs in the general population in the U.S., and increasingly worldwide, we now see more
patients reporting angina with little or no evidence of obstructive coronary artery disease based on angiographic diagnostics.
In the past 10 years, the number of ST-Elevation Myocardial Infarction patients has fallen by 50%, bypass surgery is down 40%,
and the use of stents has been reduced by 30%. We believe that this trend away from mechanical revascularization will potentially
increase the opportunity for Generx medical revascularization. The
most recently FDA approved anti-anginal drug with a novel mechanism of action is Ranexa® (ranolazine). It was FDA approved
in 2006 as a treatment for chronic angina as a metabolic modulator designed to reduce the heart’s oxygen demand. Following
FDA approval, Ranexa was acquired by Gilead Sciences for $1.4 billion in 2009. Ranexa is prescribed to be taken twice daily, generally
as a 1000 mg oral tablet and Ranolazine is now available in generic form. To
support our go to market strategy, we conducted a survey of U.S. interventional cardiologists to gauge their experience-based
assessment of the prevalence of refractory angina patients, and their openness to integrate the use of the Generx angiogenic gene
therapy product candidate, upon FDA approval, into their clinical practice. The survey confirmed that all survey responders see
patients with long-term refractory angina, and all were strongly positive and without reservation about adoption of Generx. All
cardiologists surveyed felt there is a current need for Generx to treat refractory angina and they would consider using Generx
in their daily practice if approved by the FDA. As shown in the following table, the Generx product candidate for medical revascularization
therapy generated statistically significant improvements in cardiac perfusion (measured using SPECT as a reduction in reversible
perfusion defect) as compared to placebo controls in both the U.S-based Phase 2 clinical study (AGENT-2), and a small confirmatory
international study (ASPIRE), and the observed improvements were similar in magnitude to those reported following mechanical revascularization. Generx
AGENT-2 and ASPIRE SPECT Data Generx
Clinical Studies and FDA Developments The
Generx FDA regulatory dossier represents one of the most extensive and advanced DNA-based clinical data platforms ever compiled.
Generx has been evaluated as a treatment for patients with refractory angina in four prior FDA-cleared, multi-center, randomized
and placebo-controlled clinical studies (AGENT 1-4, Phase 1/2 to Phase 2b/3) and one small international study (ASPIRE). The In
these multiple prior clinical studies, the Generx product candidate appeared safe and well-tolerated, and has generated preliminary
findings of efficacy in men and women, in measures of cardiac perfusion, cardiac performance, and angina status, including: (1)
significant improvement in exercise duration by Exercise Treadmill Testing
●
Angionetics,
Inc., an 85% owned subsidiary focused on the late-stage clinical development and commercialization of Generx, an angiogenic
●
Activation
Therapeutics, Inc., a wholly owned subsidiary focused on the development and commercialization of Excellagen®, a patented
U.S. FDA-cleared wound conforming matrix for advanced wound care; and
●
LifeAgain
Insurance Solutions, Inc., a wholly owned subsidiary focused on advanced medical data analytics for developing innovative
insurance and healthcare solutions. -4- -5- 
-6- 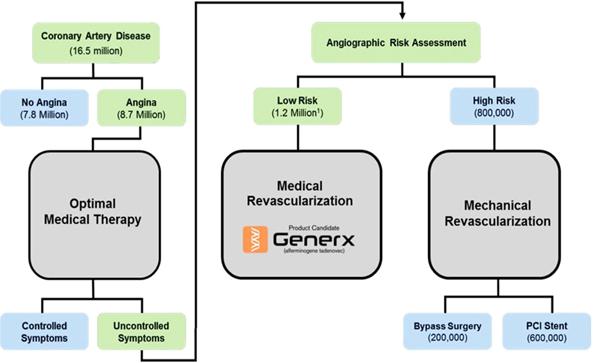
(1) Range
0.6M – 1.8M [mean 1.2M] McGillion et al., Canadian J Cardiology 28:S20-S41 (2012)
other figures, Benjamin et al., Circulation, American Heart Association, Statistics 2017. -7- 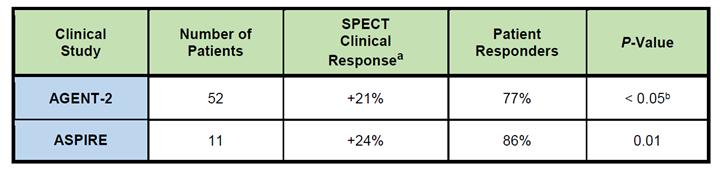
a. Improvement
in RPDS as measured by SPECT imaging at 8 weeks following a single treatment.
b. Grines
et al. JACC 42:1339-47 (2003). Tables 1 and 2. cell permeability that is believed to be activated using nitroglycerin.
| -8- |
On February 3, 2017, the FDA granted the Phase 3 AFFIRM clinical study Fast Track designation. By granting Fast Track designation to the Generx Phase 3 clinical development program, FDA acknowledges that there remains unmet medical need for patients with refractory angina. The limited available therapies for patients with refractory angina primarily address the symptoms of refractory angina by reducing myocardial oxygen demand or transiently increasing blood flow to the ischemic myocardium and require prolonged use or numerous rounds of therapy. Furthermore, available therapies have modest and heterogenous response rates. Generx is unique in its angiogenic biological mechanism of action and disease-modifying potential.
In July 2020, we submitted a protocol amendment to FDA, refining some of the patient inclusion criteria and clarifying ETT stopping criteria for enrolled patients. In addition, an adaptive trial design was incorporated to allow for interim analysis and re-estimation of sample size required to achieve the primary efficacy endpoint of statistically significant improvement in ETT with Generx compared to Placebo at 6 months. Based on further statistical analysis of historical ETT data, the target sample size was reduced from 320 patients, without an interim analysis, to 160 patients with an interim analysis after 80 patients have been enrolled. The adaptive design allows for an increase in sample size up to 226 total patients if needed to reach statistical significance.
On a global basis, over 650 patients have been enrolled in four FDA-cleared clinical studies of Generx at over 100 medical centers in the U.S., Western Europe, and Asia, 455 of whom received a one-time intracoronary administration of Generx. Based on these studies, and other pre-clinical and further international clinical evaluations, our Generx product candidate appears to be safe and well-tolerated and has generated preliminary efficacy findings in men and women, based on multiple efficacy measures within patient subset groups. Long-term safety follow-up has generated over 2,500 patient years of safety data. With the successful completion of the planned AFFIRM Phase 3 clinical study, the Generx clinical research will have evaluated over 800 patients in clinical study protocols. Based on our FDA Fast-Track designation, and our established manufacturing processes, we believe that we would be in a position to initiate the submission to the FDA of a rolling Biologics License Application (“BLA”).
FDA Registration Pathway
For
registration purposes, the FDA has classified our Generx product candidate to be an “anti-anginal” medication as a
treatment for patients who have been diagnosed with stable exertional angina due to coronary artery disease and who are no longer
responsive to current pharmaceutical therapy and mechanical interventional therapy. FDA approval of anti-anginal drugs and biologicals
requires statistically significant efficacy improvements in exercise capacity as measured by ETT compared to a placebo control
group. Developing a new and innovative anti-anginal is a challenging process and FDA approvals have been few and far between.
In the
In 2006, following a 21-year clinical and commercial development process, the FDA approved Ranexa (ranolazine), a small molecule drug in tablet form that is taken twice daily with a new mechanism of action described as metabolic modulation, to reduce the heart’s oxygen demand. Based on the Ranexa package insert, the CARISA clinical study showed that Ranexa was safe and well tolerated by refractory angina patients and that patients treated with Ranexa showed an improvement in the primary efficacy endpoint ETT of +24 seconds (+28%) compared to the placebo control over the 12- week study period. Based on our retrospective subset analysis of data from the Generx AGENT-3 clinical study, and the FDA-cleared Ad5FGF-4 Phase 3 AFFIRM clinical study design, the Generx product candidate offers the potential to meet or exceed the ETT efficacy data reported in the Ranexa CARISA clinical study. As a result, we plan to submit a BLA following successful completion of the Phase 3 AFFIRM study.
| -9- |
Generx Competitive Advantage
We believe that the most significant factors in the field of new drugs and biologics are safety and efficacy as well as relative cost, and ease of administration as compared to other products, product candidates or approaches that may be useful for treating a particular disease condition. We believe that our Generx product candidate competes favorably against the current standard of care in each of these areas:
| ● | Safety. The FDA-cleared Phase 3 AFFIRM study is preceded in the U.S. by four completed and one early discontinued study. On a global basis, over 650 patients have been enrolled in FDA-approved studies, 455 of whom received a one-time intracoronary administration of Generx, which has consistently been found to be safe and well-tolerated (based on over 2,500 patient years of safety data). Efficient uptake in the heart following intracoronary administration of Generx has been demonstrated in preclinical studies (~98% first pass extraction) and clinical studies (~90% first pass extraction). Administration of Ad5FGF-4 after stent implantation in a preclinical model of atherosclerosis and hypercholesterolemia found no evidence of increased neointima formation (restenosis) with both bare metal and drug-eluting stents. Fever is an expected side effect of adenoviral gene therapy and has been observed in ~8% of patients receiving Ad5FGF-4, occurring within the first few days after study product administration and resolving with no treatment or with antipyretic medication. No other adverse events have been associated with intracoronary administration Ad5FGF-4. | |
| ● | Effectiveness. A central finding from the Generx AGENT clinical development program is that cardiac ischemia drives Generx transfection into heart cells, and that regional cardiac ischemia is an essential precursor to support the growth of collateral blood vessels for treatment response to Generx angiogenic gene therapy. Our delivery strategy is to distribute Ad5FGF-4 throughout the microvascular circulation of the heart under conditions of transient ischemia to enhance uptake, with the angiogenic response being selective to ischemic zones. An angiogenic response to Generx has been demonstrated in preclinical studies, in which increased regional myocardial blood flow was identified by contrast echocardiography and correlated with increased vessel number, determined histologically. In clinical studies SPECT imaging has demonstrated cardiac perfusion improvements approximately up to 75% of the perfusion levels achieved from classic mechanical revascularization. The clinical response is observed in patients within four to eight weeks following administration, and it is anticipated that once formed, new vessels will persist as long as there is blood flow through the vessel. | |
| ● | Cost-Effective Manufacture. We have established and validated the Generx cGMP (defined below) manufacturing process, which is not expected to require significant additional capital investment or major process modifications for commercial manufacture. Product stability enables manufacture in large, cost-effective batch sizes. Based on our established manufacturing process, we are in a position to competitively price our Generx product candidate in alignment with cardiac stents. | |
| ● | Fits within Current Medical Practice. Generx therapy is designed to easily fit within the current practice of medicine, as a ready-to-use, one-time treatment, administered by interventional cardiologists during an approximately one-hour, out-patient, angiogram-like procedure. There are approximately 1.0 million angiogram procedures performed in the U.S. each year. Through our extensive clinical efforts, we have established appropriate dose levels, enhanced delivery techniques and simplified product administration. With regulatory approval, Generx could be the first FDA-approved gene therapy for an otherwise healthy population that would be universally affordable within healthcare medical reimbursement programs and for private pay environments. |
Additional Indications
Following our planned initial registration for refractory angina there are other potential ischemia-related cardiovascular and cerebral therapeutic opportunities that we may consider advancing forward with based on our angiogenic technology platform using varying dose levels and differing routes of administration.
Potential Pipeline of Generx (Ad5FGF-4) Medical Indications
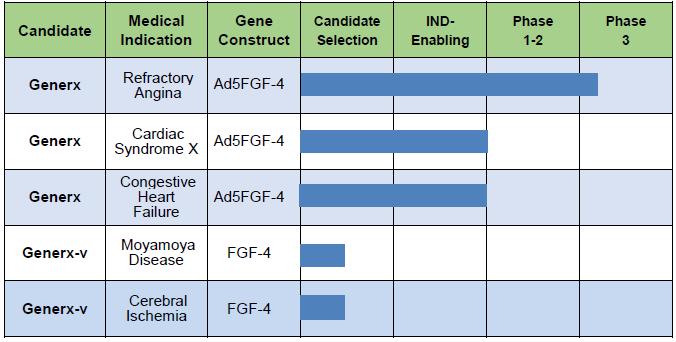
| -10- |
Cardiac Syndrome X. A meta-analysis study [Vermeltfoort et al., Clinical Research in Cardiology. 2010; 99:475-81] reported that approximately 20% of patients who have a coronary angiography due to ongoing angina do not have obvious large vessel disease, a condition generally referred to as Cardiac Syndrome X (“CSX”). Patients with CSX are presumed to have coronary disease that is diffuse and/or affects smaller vessels within the heart. CSX is therefore sometimes referred to as “microvascular angina”. CSX cannot be addressed using traditional surgical approaches such CABG or PCI. We believe patients with CSX may potentially benefit from Generx microvascular angiogenic gene therapy, and plan to conduct a U.S.-based safety and efficacy study under the current FDA-approved IND. There are approximately 200,000 patients in the U.S. with CSX, 65% of whom are women.
Congestive Heart Failure. Congestive Heart Failure is a clinical syndrome that occurs when the heart is unable to pump sufficiently to maintain blood flow to meet the body’s needs. Common causes of heart failure include coronary artery disease, heart attack, high blood pressure, atrial fibrillation, valvular heart disease, excess alcohol use, infection, and cardiomyopathy of an unknown cause. In prior clinical studies of Generx in patients with myocardial ischemia and refractory angina, approximately 50% of enrolled patients were also diagnosed with mild congestive heart failure. The rationale supporting the application of angiogenic therapy for heart failure is based on the fact that mild and/or intermittent ischemia in the sub-endocardium (inner wall) can and often does occur in congestive heart failure with almost all primary causes. In a preclinical model of heart failure due to chronic sub-endocardial ischemia, a single administration of Generx resulted in significant improvement in cardiac function [McKirnan et al., Cardiac Vascular Regeneration. 2000; 1:11-21]. These preclinical findings support the potential use of Generx [Ad5FGF-4] angiogenic gene therapy as a non-surgical treatment option for heart failure. We are evaluating a Phase 2 clinical study of Generx angiogenic therapy for the treatment of patients with certain forms of congestive heart failure due to ischemic cardiomyopathy.
Moyamoya Disease & Cerebral Ischemia. Moyamoya disease (“MMD”) is a chronic occlusive, cerebrovascular disease that is characterized by progressive stenosis at the terminal portion of the internal carotid artery and an abnormal network of collateral vessels at the base of the brain. Pursuant to the Orphan Drug Act of 1983, MMD is an orphan indication, with <1 case per 100,000 in the U.S. The prevalence of MMD is much higher in East Asian countries than in Western countries. The highest prevalence of MMD is found in Japan at 3.16 per 100,000. Currently, there is no known medical treatment capable of reversing or stabilizing progression of MMD. Surgical revascularization such as extracranial-intracranial bypass is the preferred procedure for MMD patients with the main goal of preventing further ischemic injury by increasing collateral blood flow to hypo-perfused areas of the cortex. Collateral vessels are seen to sprout from bypassed vessels, thus providing increased blood flow to ischemic regions of the brain. We believe that Generx may potentially offer a new and simpler medical revascularization approach to the treatment of MMD, with a view toward further clinical development of angiogenic gene therapeutics for patients with a broader range of cerebral ischemic conditions, including vascular dementia. Preclinical studies have demonstrated that adenovectors can transfect cells in the brain, and we are investigating potential routes of administration to MMD patients that include, (1) adjunctive application of Ad5FGF-4 during burr hole surgery to augment collateralization, and (2) infusion into the carotid artery, to target ischemic regions and stimulate collateral vessel formation.
Angiogenic Research Initiative for COVID-19.
Early research has provided evidence of respiratory, neurological, and cardiac abnormalities in patients who have had severe COVID-19 immunological response requiring acute care (including protracted hospitalization and the need for mechanical ventilation). For patients who have survived and seek to return to normal life, several continuing residual adverse medical conditions appear to persist.
While the scientific literature remains uncertain, it has been suggested that mechanisms by which COVID-19 could lead to cardiovascular morbidity include direct myocardial injury as a result of inflammatory cascade or cytokine release, acute coronary syndrome from acute inflammation-triggered destabilization of atheroma, microvascular damage due to disseminated intravascular coagulation and thrombosis, direct entry of SARS-CoV-2 into myocardial cells via ACE2 receptors, and hypoxemia combined with metabolic demands of acute illness leading to myocardial injury akin to a myocardial infarction.
Based
on these preliminary insights, Gene Biotherapeutics’ research is focused on the design of an observational clinical study
to evaluate if COVID-19 may exacerbate microvascular damage and perfusion impairment in patients with pre-existing coronary artery
disease and cardiac reversible perfusion defects (“RPD”) prior to COVID-19 infection. We are proposing to assess
the damage using SPECT (Single-Photon Emission Computed Tomography) imaging to evaluate changes in
| -11- |
Commercialization Business Strategy
We
are committed to applying our first-mover scientific and clinical development leadership position in the field of angiogenic gene
therapy for the treatment of patients with a variety of cardiovascular conditions which are related by insufficient cardiac perfusion
and other potential ischemia-related cerebral therapeutic opportunities as well as advanced tissue engineering applications. The
core elements of our
| ● | Following U.S. registration for refractory angina, initiate the registration process to market and sell Generx in China, the Russian Federation, and the CIS with our current strategic partners, and consider registration in other prioritized regional markets; | |
| ● | Following FDA approval, we would also plan to (1) enter a strategic agreement(s) to market and sell Generx in other countries worldwide, or (2) undertake a terminal value transaction covering the sale of Generx to an established strategic player which has established worldwide marketing, sales, and distribution capabilities; | |
| ● | Expand the initial
labeling of Generx by initiating a Phase 2 clinical study to support the use of Generx for patients with CSX, which
is characterized by symptomatic angina in the absence of large coronary artery obstruction, and for certain forms | |
| ● | Advance our pre-clinical
research which is focused on applying our Ad5FGF-4 technology platform as a potential treatment for patients with MMD,
an orphan medical |
| ||
| ● | Establish a Generx
patient registry and conduct additional clinical studies to evaluate the safety and clinical efficacy of repeat dosing of
Generx in patients as their coronary artery disease advances causing additional perfusion defects |
| ||
| ● | Initiate additional studies to assess the potential long-term prognostic benefits of refractory angina patients receiving angiogenic therapy through medical revascularization. |
Government Regulation
Gene
therapy biologics are subject to extensive regulation in the United States under the federal Food, Drug, and Cosmetic Act. In
addition, biologics are also regulated under the Public Health Service Act. Both statutes and their corresponding regulations
govern, among other things, the testing, manufacturing, distribution, safety, efficacy, labeling, storage, record keeping, advertising
and other promotional practices involving biologics or new drugs. FDA approval or other clearances must be obtained before clinical
testing, and before manufacturing and marketing
Any
product candidate we develop will require regulatory approvals on a country-by-country basis before human trials and additional
regulatory approvals before marketing. The FDA receives reports on the progress of each phase of testing, and it may require the modification, suspension,
or termination of trials if an unwarranted risk is present to patients. If the FDA imposes a clinical hold, trials may not recommence
without FDA authorization and then only under terms authorized by the FDA. The IND application process can thus result in substantial
delay and expense.
| -12- |
Our
Generx product candidate is a gene therapy product, which is a relatively new category of therapeutics.
After
the completion of trials of a new drug or biologic product, we will have to secure FDA marketing approval. The New Drug Application
(“NDA”) or BLA must include results of product development, laboratory, animal and human studies, and
manufacturing information. The testing and approval processes require substantial time and effort and there can be
Notwithstanding
the submission of all relevant data, the FDA may ultimately decide that the NDA or BLA does not satisfy its regulatory criteria
for approval and may require additional
In
addition to FDA approval for the commercialization of our product candidates, our business is subject to state and federal laws
regarding environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy
and Recovery Act and the Toxic Substances Control Act
To
the extent that we conduct operations outside the United States, any such operations would be similarly regulated by various agencies
and entities in the countries in which we operate. The regulations of these countries may conflict with those in the United States
and may vary from country to country. In markets outside the United States, we may be required to obtain approvals, licenses,
or certifications from a country
Competition
The
pharmaceutical industry is intensely competitive. Our
Our
Generx
| -13- |
We
are aware of products currently
Ranexa |
| ● | The Neovasc Reducer™ (“Reducer”) is a stainless steel, hourglass-shaped medical device that is implanted into the coronary sinus using a procedure similar to that used for stent implantation. It is designed to create a focal narrowing in the coronary sinus, resulting in increased back pressure and redistribution of blood into ischemic myocardium. In 2015, results from a Phase 2 study (the “COSIRA” study; N=104) were published, reporting that significantly more patients in the treatment group, as compared to control, had an improvement in CCS class and quality of life at 6 months, but no significant improvement in exercise time. In December 2018, Neovasc announced publication of 12-year follow-up data from 7 patients demonstrating sustained improvement of angina class compared with baseline status. The Reducer is currently available only in the European Union, receiving CE mark designation in 2011. In October 2018, Neovasc announced that the Reducer™ was granted Breakthrough Device designation by the U.S. FDA, and in December 2019, Neovasc announced submission to FDA of a Premarket Approval application (PMA) for the treatment of refractory angina. On October 27, 2020, an 18 member FDA Advisory Committee reviewed the PMA submission, voting 17 to 1 “against” on the issue of a reasonable assurance of effectiveness, voting 14 to 4 “in favor” that the Reducer is safe when used as intended, and voting 13 to 3 “against” (2 abstained) on whether the relative benefits outweighed the relative risks. | |
| ● | Caladrius Biosciences is developing an autologous CD34+ stem cell product candidate for refractory angina (“CLBS14”). Caladrius acquired an exclusive worldwide license to data and regulatory filings for the late stage CD34+ cell therapy program from Shire plc in March 2018. CD34+ therapy is thought to work by increasing microvascular blood flow in the heart muscle via the development and formation of new blood vessels. Cells are collected from patients after drug-induced mobilization, followed by isolation, concentration, and formulation prior to intramyocardial injection guided by mapping catheter (NOGA). CLBS14 has been studied in Phase 1, Phase 2 and Phase 3 randomized, double-blind placebo-controlled clinical trials that reveal significant improvements in exercise capacity and angina frequency. According to public records, initiation of a Phase 3 confirmatory trial is postponed pending access to sufficient capital to complete the study uninterrupted. | |
| In May 2020, Caladrius announced positive results from a 20-patient Phase 2 proof of concept study with CD34+ cell therapy (CLBS16) in patients with CSX. Data showed statistically significant improvement in coronary flow reserve correlating with symptom relief after a single intracoronary injection of CLBS16. | ||
| ● | XyloCor Therapeutics is developing an adenovirus-based gene therapy encoding a hybrid gene for human vascular endothelial growth factor (“XC001”) for patients with refractory angina. XC001 is designed to relieve angina by promoting angiogenesis. In July 2020, XyloCor announced dosing of the first patients in the initial Phase 1/2 dose-escalation clinical study. XC001 is administered by transthoracic epicardial injection. | |
| ● | BioCardia Inc. is developing the CardiAmp™ Cell Therapy System, which provides an autologous bone marrow-derived stem cell therapy for the treatment of chronic myocardial ischemia. In July 2020, BioCardia announced activation of a Phase 3 clinical trial studying percutaneously injected cells for the treatment of refractory angina and chronic myocardial ischemia. | |
| ● | Juventas
Therapeutics is developing a non-viral, plasmid gene therapy product candidate (JVS-100) that expresses stromal cell-derived
factor-1 (“SDF-1”) for the treatment of advanced ischemic heart failure. SDF-1 has been shown to create
a homing signal that recruits the body |
| Manufacturing
Strategy
|
We
will rely on contract manufacturing for the Generx product candidate. Based on the FDA clearance of the Generx Phase 3 clinical
study protocol, all significant
| -14- |
We
have been actively advancing our Generx product candidate’s engineering and process technology in preparation for commercialization.
The adenovector Ad5FGF-4 is propagated in suspension cultures of fully characterized HEK 293 cells using serum-
Generx’s
long-term product stability (at the current storage temperature of -70
In
March 2021, the Company entered into an agreement with FUJIFILM Diosynth Biotechnologies (“FDB”) to manufacture the
Generx [Ad5FGF-4] angiogenic gene therapy product candidate for Phase 3 clinical evaluation for the treatment of refractory angina
due to late-stage coronary artery disease. Manufacturing operations will be conducted at FDB’s facilities in College Station,
Texas where FDB will perform technology transfer and process development activities for Phase
Marketing and Sales
Our
product candidates, such as Generx, must undergo clinical trials before any marketing and sales can begin. If we should obtain
marketing approvals, we do not currently have the financial resources and internal capabilities to market and sell Generx. Commercialization
Relationships Huapont
Life Sciences Co. Ltd (“Huapont”). Huapont is a China-based company focused on the research and development of
new and innovative healthcare products, and the manufacture, marketing and sale of leading pharmaceutical products, active pharmaceutical
ingredients, and a portfolio of safe and effective agricultural herbicides serving the agricultural business throughout the U.S.
and South American markets. Huapont’s pharmaceutical business includes dermatology products, cardiovascular products, anti-tuberculosis
agents, autoimmune-related products, and oncology-related products. Huapont’s API business involves the production and sale
of bulk pharmaceutical chemicals, pharmaceutical intermediates, and preparations of Western medicines, with current annual revenues
of approximately U.S. $1.5 billion, and approximately 12,000 employees operating throughout Mainland China. Huapont is listed
on the Shenzhen Stock Exchange (002004.SZ) and carries a current market capitalization of approximately U.S. $1.7 billion. In
July 2016, Pineworld Capital Limited, an investment fund affiliated with Huapont acquired a 15% preferred stock equity interest
in our Angionetics, Inc. subsidiary (the entity that holds the Generx product) in exchange for a $3.0 million investment. Concurrently
with that investment, Angionetics entered into a Distribution and License Agreement, granting Huapont an exclusive license to
clinically develop, manufacture, market and sell the Generx angiogenic gene therapy product candidate in mainland China. The distribution
and license rights commence only after we obtain U.S. FDA approval for marketing and sale of Generx in the United States. Once
the license is effective, Huapont has agreed, at its expense, to use commercially reasonable efforts to conduct clinical trials,
make regulatory filings and take such other actions as may be necessary to commercialize Generx in mainland China. The Distribution
and License Agreement calls for Huapont to make quarterly royalty payments at a rate of 10% of net sales of Generx products in
mainland China, reducing to a 5% royalty based on the volume of annual sales. The royalty payments commence on the first commercial
sale and expire on the earlier of the termination of any patent or regulatory exclusivity in China or fifteen years after the
first commercial sale. The term of the agreement continues (unless terminated for breach) until Huapont has no remaining payment
obligations to Angionetics. Upon expiration (but not an earlier termination) Huapont shall have a perpetual, non-exclusive, fully
paid-up, and royalty-free license to Generx in mainland China. Olaregen
Therapeutix Inc. In July 2018, we sold our Excellagen product to Olaregen for aggregate consideration of up to $4,000,000.
At closing, we received a cash payment of $650,000, and we will be entitled to receive royalty payments of 10% of worldwide net
sales
| -15- |
On
April 10, 2020, our Activation Therapeutics, Inc. subsidiary entered into a License and Patent Assignment Agreement with Shanxi
(the “Shanxi Assignment Agreement”) pursuant to which we transferred of all of our residual rights and assets related
to our Excellagen product to Shanxi. Under the terms of the Shanxi Assignment Agreement, we transferred all our license rights
to manufacture, use, market and sell Excellagen to
Nostrum Pharmaceuticals,
LLC. In May 2020, after the period covered by this report, we entered into a Preferred Stock Purchase Agreement with
Nostrum selling 1,700,000 shares of our newly authorized Series B Convertible Preferred Stock in exchange for $1,700,000. The
shares of Series B Convertible Preferred Stock are convertible into an aggregate of 150,442,478 shares of Common Stock. In
addition, Nostrum entered into an agreement with the holder of our outstanding Series A Convertible Preferred Stock, under
which Nostrum purchased 220 shares of our Series A Convertible Preferred Stock, convertible into an aggregate of 19,469,026
shares of Common Stock and agreed to purchase up to 570 additional Series A Convertible Preferred Stock. Since May 2020, such
holder has converted 397 shares of Series A Convertible Preferred Stock into 35,132,755 shares of our Common
stock, that has increased our outstanding Common Stock to 49,622,154 shares as of March 31, 2021. As a result
of these transactions, Nostrum currently controls approximately 75.2% of the voting interests of our Company. Nostrum
is a privately held pharmaceutical company engaged in the formulation and commercialization of specialty pharmaceutical
products and controlled release, orally administered, branded and generic drug products. We believe that Nostrum’s
assets and experience in the formulation and commercialization of pharmaceutical products will facilitate the administration
and completion of the AFFIRM Phase 3 clinical trial on a cost-effective basis. However, we do not have
Intellectual Property and Licensing-
In
connection with the Schering portfolio, we acquired the rights to certain patents owned by the University of California related
to the use of the catheter as part of
In
June 2016 we entered into a Distribution and License Agreement with an affiliate of Huapont whereby we granted the Huapont affiliate
an exclusive license to clinically develop
| -16- |
In
July 2018, we sold our
As
of December 31, 2019, we had Available
Information Our
website address is www.genebiotherapeutics.com. We make available, free of charge, through our website our Annual Report on Form
10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports filed or furnished pursuant
to Section You
should carefully review and consider the risks described below, as well as the other information in this report and in other reports
and documents we file with the SEC when evaluating our business and future prospects. The risks and uncertainties described below
are not the only ones we face. Additional risks and uncertainties, not presently known to us, or that we currently perceive as
immaterial or remote, may also occur. If any of the following risks or any additional risks and uncertainties actually occur,
our business could be materially harmed, and our financial condition, results of operations and future growth prospects could
be materially and adversely affected. In that event, the market price of our Risks
Related to the Development of Product Candidates The
regulatory approval processes of the FDA are inherently unpredictable, and if we are ultimately unable to obtain regulatory approval
for our product candidates, we may never generate revenue or achieve profitability. To
generate revenues, we must successfully complete clinical trials of our product candidates Generally,
there is a high rate of failure for drug candidates proceeding through clinical trials. We
cannot be certain that any of our product candidates will be successful in clinical trials or receive regulatory approval. Further,
our product candidates may not receive regulatory approval even if
ITEM
1A. RISK
FACTORS
●
The
FDA may disagree with the design or implementation of our clinical trials;
●
We
may be unable to demonstrate sufficiently to the FDA that our product candidate is safe
●
The
results of our clinical trials may not meet the level of statistical significance required by the FDA for approval; -17-
●
The
approval policies or regulations of the FDA may change significantly, in a manner rendering our clinical data insufficient
for approval. on the performance of costly post-marketing clinical trials, may approve a product candidate with a label that does not include
the labeling claims necessary or desirable for the successful commercialization of that product candidate or may restrict its
distribution. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
Clinical trials are expensive, time-consuming, and difficult to design and implement, and involve an uncertain outcome.
Before obtaining marketing approval from the FDA or other comparable foreign regulatory authorities for the sale of our product candidates, we must complete pre-clinical development and extensive clinical trials to demonstrate the safety and efficacy of our product candidates. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Although we are planning for certain clinical trials relating to Generx and our other product candidates, there can be no assurance that the FDA will accept our proposed trial designs.
We may experience delays in our clinical trials, and we do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| ● | the FDA disagreeing as to the design or implementation of our clinical studies; | |
| ● | reaching mutually acceptable agreements with prospective contract research organizations (“CROs”); | |
| ● | securing a sufficient number of clinical trial sites on acceptable terms; | |
| ● | clinical sites deviating from trial protocol or dropping out of a trial; | |
| ● | obtaining institutional review board (“IRB”), approval at each site, or independent ethics committee, approval at any sites outside the United States; | |
| ● | securing sufficient quantities of our product candidate from third party contract manufacturers to support the trial; | |
| ● | any changes to our manufacturing process that may be necessary or desired; | |
| ● | addressing patient safety concerns that arise during the course of a trial; | |
| ● | imposition of a clinical hold by regulatory authorities, including as a result of unforeseen safety issues or side effects or failure of trial sites to adhere to regulatory requirements; | |
| ● | the occurrence of serious adverse events in trials of the same class of agents conducted by other companies or institutions; | |
| ● | changes to clinical trial protocols; |
| -18- |
| ● | lack of adequate funding to continue the clinical trial. |
If the third parties that we rely on for pre-clinical and clinical trial support do not successfully perform their contractual legal and regulatory duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We
have relied upon and plan to continue to rely upon third-party medical institutions, clinical investigators, contract laboratories
and other third party CROs to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties
Regulatory
authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators, and trial sites. If we
or any of our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials In
addition, our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot
control whether or not they devote sufficient time and resources to our on-going clinical, non-clinical and preclinical programs.
If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced
or if the quality or accuracy of the clinical data, they obtain is compromised due to the failure to adhere to our clinical protocols,
regulatory requirements or for other reasons, our clinical trials may be extended, delayed, or terminated and we may not be able
to obtain regulatory approval for or successfully commercialize our product candidates If
any If
we are unable to enroll patients in our clinical trials, our research and development efforts could be adversely affected. The
timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll
a sufficient number of patients who remain in the study until its conclusion. We may experience difficulties in patient enrollment
in our clinical trials for a variety of reasons. Patient enrollment is affected by many factors including: Many
pharmaceutical companies are conducting clinical trials in patients with the disease indications that our potential drug products
target. As a result, we must compete with them for clinical sites, physicians and the limited number of patients who fulfill the
stringent requirements for participation in clinical trials. Also, due to the confidential nature of clinical trials, we do not
We
may be unable to maintain sufficient clinical trial liability insurance to fully insure against liabilities arising out of clinical
We
will require all patients enrolled in our clinical trials to sign consents, which explain various risks involved with participating
in the trial. However, patient consents provide only a limited level of protection, and it may be alleged that the consent did
not address or did not adequately address a risk that the patient suffered from. Additionally, we will generally be required to
indemnify the clinical product manufacturers, clinical trial centers, medical professionals and other parties conducting related
activities in connection with losses they may incur through their involvement in the clinical trials. We may not be able to obtain
or maintain product liability insurance on acceptable terms or with adequate coverage against potential liabilities. Our
inability to retain sufficient clinical trial liability insurance at an acceptable cost to protect against potential liability
claims could prevent or inhibit our ability to conduct clinical trials for product candidates we develop. We may be unable to
obtain appropriate levels of such insurance. Even if we do secure clinical trial liability insurance for our programs, we may
not be able to achieve sufficient levels of such insurance. Any claim that may be brought against us could result in a court judgment
or settlement in an amount that is not covered, in whole or in part, by our insurance or that is more than the limits of our insurance
coverage. We expect we will supplement our clinical trial coverage with product liability coverage in connection with the commercial
launch of Generx or other product candidates we develop in the future; however, we may be unable to obtain such increased coverage
on acceptable terms or at all. If we are found liable in a clinical trial lawsuit or a product liability lawsuit in the future,
we will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that
are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. We
currently have only one significant product candidate—our Generx product candidate—and our business is substantially
dependent on its success. We
do not currently have any viable product candidates other than Generx. Accordingly, our success is substantially dependent on
our ability to successfully secure marketing approval and to commercialize Generx. If we fail to secure marketing approval for
Generx, we could be forced to try to secure an alternative product candidate. Our internal research and development capabilities
are limited and will initially be focused on the Phase 3 Generx clinical trial. We may evaluate, acquire, license, develop and/or
market additional product candidates and technologies. We do not currently have substantial resources to procure additional technologies.
The success of this strategy depends partly upon our ability to identify, select, and acquire promising pharmaceutical product
candidates and products. The process of proposing, negotiating, and implementing a license or acquisition of a product candidate
or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing and
sales resources, may compete with us for the license or acquisition of product candidates and approved products. We have limited
resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate
them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities
that are never completed, or we may fail to realize the anticipated benefits of such efforts. We may not be able to acquire the
rights to additional product candidates on terms that we find acceptable, or at all. If we are unable to receive marketing approval
and successfully commercialize Generx we may not be able to secure rights to another viable product candidate and may be forced
to cease operations. Interim
“top-line” and preliminary data from our clinical trials may change as more patient data become available and are
subject to verification procedures that could result in material changes in the final data. From
time to time, we may publicly disclose interim top-line or preliminary data from our clinical trials, which is based on a preliminary
analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive
review of the data related to the particular study or trial. We also make assumptions, estimations, calculations, and conclusions
as part of our analyses of data, and we may not have received or had the opportunity to evaluate all data fully and carefully.
As a result, the top-line, or preliminary results that we report may differ from future results of the same studies, or different
conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Top-line or
preliminary data also remain subject to verification procedures that may result in the final data being materially different from
the top-line or preliminary data we previously published. As a result, top-line and preliminary data should be viewed with caution
until the final data are available. Regulatory
agencies may not accept or agree with our assumptions, estimates, calculations, conclusions, or analyses or may interpret or weigh
the importance of data differently, which could impact the value of the particular program, the approvability or commercialization
of the particular product candidate or product and our company in general. In addition, the information we choose to publicly
disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others
may not agree with what we determine is material or otherwise appropriate information to include in our disclosure. If
the interim, top-line or preliminary data that we report differ from actual results, or if others, including regulatory authorities,
disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed,
which could harm our business, operating results, prospects, or financial condition. We
have obtained Fast Track Designation for Generx, but that designation may not lead to a faster development, regulatory review,
or approval. If
a product is intended for the treatment of a serious condition and nonclinical or clinical data demonstrate the potential to address
unmet medical need for this condition, a product sponsor may apply for FDA Fast Track designation. We have obtained Fast Track
designation for Generx for investigation into the treatment of refractory angina, providing opportunity for expedited clinical
development and regulatory review. Fast Track Designation does not ensure that we will receive marketing approval or that approval
will be granted within any particular timeframe. We may not experience a faster development or regulatory review or approval process
with Fast Track designation compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation
if it believes that the designation is no longer supported by data from our clinical development program. Fast Track designation
alone does not guarantee qualification for the FDA’s priority review procedures. If
the FDA does not conclude that our product candidates satisfy the requirements for the 505(b)(2) regulatory approval pathway,
or if the requirements for approval of any of our product candidates under Section 505(b)(2) are not as we expect, the approval
pathway for our product candidates will likely take significantly longer, cost significantly more, and encounter significantly
greater complications and risks than anticipated, and in any case may not be successful. We
intend to seek FDA approval through the 505(b)(2) regulatory pathways for Generx. Section 505(b)(2) of the Food Drug and Cosmetics
Act permits the filing of an NDA where at least some of the information required for approval comes from studies that were not
conducted by or for the applicant. If the FDA does not allow us to pursue the 505(b)(2) regulatory pathways for our product candidates
as anticipated, we may need to conduct additional clinical trials, provide additional data and information, and meet additional
standards for regulatory approval. If this were to occur, the time and financial resources required to obtain FDA approval for
our product candidates would likely substantially increase. Moreover, the inability to pursue the 505(b)(2) regulatory pathways
could result in new competitive products reaching the market faster than our product candidates, which could materially adversely
impact our competitive position and prospects. Even if we can pursue the 505(b)(2) regulatory pathways for a product candidate,
we cannot assure you that we will receive the requisite or timely approvals for commercialization of such product candidate. In
addition, we expect that our competitors will file citizens’ petitions with the FDA in an effort to persuade the FDA that
our product candidates, or the clinical studies that support their approval, contain deficiencies. Such actions by our competitors
could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2). Our
product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval,
limit the commercial profile of an approved label, or result in other significant negative consequences. Undesirable
side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials
and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign
authorities. The clinical evaluation of Generx and our other product candidates in patients is still in the early stages and it
is possible that there may be side effects associated with their use. Results of our trials could reveal a high and unacceptable
severity and prevalence of side effects. In such an event, we, the FDA, the IRBs at the institutions in which our studies are
conducted, or the Data Safety Monitoring Board could suspend or terminate our clinical trials, or the FDA or comparable foreign
regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted
indications. While
we are not presently aware of any side effects from the use of Generx, possible serious side effects of gene transfer include
viral or gene product toxicity resulting in inflammation or other injury to the heart or other parts of the body. The development
or worsening of cancer in a patient could potentially be a perceived or actual side effect of gene therapy technologies. Furthermore,
there is a possibility of side effects or decreased effectiveness associated with an immune response toward any viral vector or
gene used in gene therapy. The possibility of such response may increase if there is a need to deliver the viral vector more than
once. Treatment-related
side effects could also affect patient recruitment or the ability of enrolled patients to complete the clinical trial or result
in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating
medical staff. We expect to have to train medical personnel using our product candidates to understand the side effect profiles
for our clinical trials and upon any commercialization of any of our product candidates. Inadequate training in recognizing or
managing the potential side effects of our product candidates could result in patient injury or death. Any of these occurrences
may harm our business, financial condition, and prospects significantly. If
we elect or are forced to suspend or terminate any planned clinical trial of Generx or any other of our product candidates, the
commercial prospects for that product will be harmed and our ability to generate product revenue from that product may be delayed
or eliminated. Furthermore, any of these events could prevent us or our partners from achieving or maintaining market acceptance
of the affected product and could substantially increase the costs of commercializing our product candidates and impair our ability
to generate revenue from the commercialization of these products. Risks
Related to Product Commercialization Even
if we obtain regulatory approvals to commercialize Generx or other product candidates, our product candidates may not be accepted
by physicians or the medical community in general. Our
ongoing business depends on the success of our technologies and product candidates. Gene-based therapy, like our Generx product
candidate, is a relatively new and rapidly evolving medical approach. Biotechnology and pharmaceutical companies have successfully
developed and commercialized only a limited number of biologic-based products and to date only a limited number of cellular and
gene therapy products have been approved by the U.S. FDA. Our product candidates, and the technology underlying them, are new
and unproven and there is no guarantee that health care providers or patients will be interested in our products even if they
are approved for use. We
cannot be certain that Generx or any other product candidate we successfully develop will be accepted by physicians, hospitals,
and other health care facilities. The degree of market acceptance of any drugs we develop depends on a number of factors, including: If
the market does not accept our products or product candidates, when and if we are able to commercialize them, then we may never
become profitable. It is difficult to predict the future growth of our business, if any, and the size of the market for our product
candidates because the market and technology are continually evolving. There can be no assurance that our technologies and product
candidates will prove superior to technologies and products that may currently be available or may become available in the future
or that our technologies or research and development activities will result in any commercially profitable products. If our products
do not gain market acceptance, we may not be able to fund future operations either through operating or financing activities. Even
if we obtain marketing approval for Generx or another product candidate, we will still face extensive and ongoing regulatory requirements
which could significantly impact our operations. Any
product candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data,
labeling, packaging, distribution, adverse event reporting, storage, recordkeeping, export, import, advertising, and promotional
activities for such product, among other things, will be subject to extensive and ongoing requirements of and review by the FDA
and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports,
establishment registration and drug listing requirements, continued compliance with cGMP requirements relating to manufacturing,
quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution
of samples to physicians and recordkeeping and GCP requirements for any clinical trials that we conduct post-approval. Even
if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for
which the product candidate may be marketed or to the conditions of approval, including a requirement to implement a REMS. If
any of our product candidates receives marketing approval, the accompanying label may limit the approved indicated use of the
product candidate, which could limit sales of the product candidate. The FDA may also impose requirements for costly post-marketing
studies or clinical trials and surveillance to monitor the safety or efficacy of a product. Violations of the Federal Food, Drug,
and Cosmetic Act, or FDCA, relating to the promotion of prescription drugs may lead to FDA enforcement actions and investigations
alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws. Later
discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes or
failure to comply with regulatory requirements, could result in: Further,
the FDA’s policies may change, and additional government regulations may be enacted that could impose extensive and ongoing
regulatory requirements and obligations on any product candidate for which we obtain marketing approval. If we are slow or unable
to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain
regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business,
prospects, and ability to achieve or sustain profitability. Healthcare
reform measures could hinder or prevent our product candidates’ commercial success. New
laws, regulations and judicial decisions, or new interpretations of existing laws, regulations, and decisions, that relate to
healthcare availability, methods of delivery or payment for products and services, or sales, marketing, or pricing, may limit
our potential revenue, and we may need to revise our research and development programs. The continuing efforts of the U.S. and
foreign governments, insurance companies, managed care organizations and other payors of health care services to contain or reduce
health care costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to
generate revenues and achieve and maintain profitability. We cannot predict the reform initiatives that may be adopted in the
future or whether initiatives that have been adopted will be repealed or modified. Further
federal and state proposals and health care reforms are likely which could limit the prices that can be charged for the product
candidates that we develop and may further limit our commercial opportunities. Our proposed products may not be considered cost-effective,
and coverage and reimbursement may not be available or sufficient to allow us to sell our proposed products on a profitable basis.
Our results of operations could be materially adversely affected by proposed healthcare reforms, by the Medicare prescription
drug coverage legislation, by the possible effect of such current or future legislation on amounts that private insurers will
pay and by other health care reforms that may be enacted or adopted in the future. We
intend to rely on third parties to produce commercial supplies of any approved product candidate, and our commercialization of
any future product could be stopped or delayed or made less profitable if third party manufacturers fail to obtain approval of
the FDA or comparable regulatory authorities or fail to provide us with drug product in sufficient quantities or at acceptable
prices. The
manufacture of biotechnology and pharmaceutical products is complex and requires significant expertise, capital investment, process
controls and know-how. Common difficulties in biotechnology and pharmaceutical manufacturing may include: We
do not currently have nor do we plan to acquire the infrastructure or capability internally to produce an adequate supply of compounds
to meet future requirements for clinical trials and commercialization of our products or to produce our products in accordance
with cGMP prescribed by the FDA. Drug manufacturing facilities are subject to inspection before the FDA will issue an approval
to market a new drug product, and all of the manufacturers that we intend to use must adhere to the cGMP regulations prescribed
by the FDA. We
expect to rely on third-party manufacturers for clinical supplies of our product candidates that we may develop. These third-party
manufacturers will be required to comply with cGMPs, and other applicable laws and regulations. We will have no control over the
ability of these third parties to comply with these requirements, or to maintain adequate quality control, quality assurance and
qualified personnel. If the FDA or any other applicable regulatory authorities do not approve the facilities of these third parties
for the manufacture of our other product candidates or any products that we may successfully develop, or if it withdraws any such
approval, or if our suppliers or contract manufacturers decide they no longer want to supply or manufacture for us, we may need
to find alternative manufacturing facilities, in which case we might not be able to identify manufacturers for clinical or commercial
supply on acceptable terms, or at all. Any of these factors would significantly impact our ability to develop, obtain regulatory
approval for or market our product candidates and adversely affect our business. Manufacturing
biologic products is subject to a multitude of manufacturing risks, any of which could substantially increase our costs and limit
supply of our products. We
and/or our third-party manufacturers may be adversely affected by developments outside of our control, and these developments
may delay or prevent further manufacturing of our products. Adverse developments may include: If
we or our third-party manufacturers were to encounter any of the above difficulties, or otherwise fail to comply with contractual
obligations, our ability to provide any product for commercial purposes would be jeopardized. This may increase the costs associated
with completing our commercial production. We may also have to take inventory write-offs and incur other charges and expenses
for products that fail to meet specifications or pass safety inspections. Inability to meet the demand for our product candidate
could damage our reputation and the reputation of our product among physicians, healthcare payors, patients, or the medical community,
which could adversely affect our ability to operate our business and our results of operations. If production difficulties cannot
be solved with acceptable costs, expenses, and timeframes, we may be forced to abandon our commercialization plans, which could
have a material adverse effect on our business, prospects, financial condition, and the value of our securities. If
we are unable to develop satisfactory sales and marketing capabilities, we may not succeed in commercializing Generx or any other
product candidate. We
have limited experience in marketing and selling drug products. Typically, pharmaceutical companies would employ groups of sales
representatives and associated sales and marketing staff numbering in the hundreds to thousands of individuals to call on many
physicians and hospitals. If we seek to market and sell our drugs directly, we will need to hire additional personnel skilled
in marketing and sales. The establishment of a direct sales force or a contract sales force or a combination direct and contract
sales force to market our products will be expensive and time-consuming and could delay any product launch. Further, we can give
no assurances that we may be able to maintain a direct and/or contract sales force for any period or that our sales efforts will
be sufficient to grow our revenues or that our sales efforts will ever lead to profits. We
may seek to collaborate with a third party to market our products. If we seek to collaborate with a third party, we cannot be
sure that a collaborative agreement can be reached on terms acceptable to us. We cannot be sure that we will be able to acquire,
or establish third party relationships to provide, any or all these marketing and sales capabilities. We
operate in a highly competitive industry and the emergence of an alternative product or technology could significantly impact
the market opportunity for our products. Biopharmaceutical
product development is highly competitive and subject to rapid and significant technological advancements. We face and will continue
to face intense competition from a variety of businesses, including large, fully integrated, well-established pharmaceutical companies
who already possess a large share of the market, specialty pharmaceutical and biopharmaceutical companies, academic institutions,
government agencies and other private and public research institutions in the United States, the European Union, and other jurisdictions.
These companies have significantly greater financial resources and expertise in research and development, manufacturing, preclinical
testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved drugs than we do. This may make it
easier for them to respond more quickly than us to new or changing opportunities, technologies, or market needs. For
our Generx product candidate, we will have to demonstrate that it provides advantages over existing standards of care including
stents, enhanced external counter-pulsation, and Ranexa® (ranolazine). In addition, a number of competitors are developing
alternative treatments for refractory angina, including product candidates being developed by Neovasc, BioCardia, Caladrius, and
others. Our
competitors may develop more effective or more affordable products or achieve earlier patent protection or product commercialization
and market penetration than us. As these competitors develop their technologies, they may develop proprietary positions that prevent
us from successfully commercializing our future products. If we are unable to adapt, products and technologies developed by our
competitors may render our products and product candidates uneconomical or obsolete, and we may not be successful in marketing
our products and product candidates against competitors. We may never be able to capture and maintain the market share necessary
for growth and profitability and there is no guarantee we will be able to compete successfully against current or future competitors. If
we successfully commercialize Generx or another product candidate, we will face the risk of product liability claims, which could
adversely affect our business and financial condition. Our
sales and marketing will expose us to product liability risks that are inherent in the testing, manufacturing, and marketing of
biotechnology products. Product liability may result from harm to patients using our products, such as a complication that was
either not communicated as a potential side effect or was more extreme than communicated. Failure to obtain or maintain sufficient
product liability insurance or otherwise protect against product liability claims could prevent or delay the commercialization
or marketing of our products or product candidates or expose us to substantial liabilities and diversions of resources, all of
which can negatively impact our business. Regardless of the merit or eventual outcome, product liability claims may result in
withdrawal of product candidates from clinical trials, costs of litigation, damage to our reputation, substantial monetary awards
to plaintiffs and decreased demand for products. Risks
Related to Intellectual Property Rights Our
intellectual property may not be sufficient to protect our products from competition, which may negatively affect our business
as well as limit our partnership or acquisition appeal. The
patents relating to the fundamental processes for our Generx product candidate have expired. We do not currently have any patent
protection related to Generx. For Generx, and other product candidates we may develop, we rely on trade secrets, know-how, continuing
technological innovations and licensing opportunities to develop and maintain our competitive position. We
may be subject to competition despite the existence of intellectual property we license or own. We can give no assurances that
our intellectual property claims will be sufficient to prevent third parties from designing around patents we own or license and
developing and commercializing competitive products. The existence of competitive products that avoid our intellectual property
could materially adversely affect our operating results and financial condition. Furthermore, limitations, or perceived limitations,
in our intellectual property may limit the interest of third parties to partner, collaborate or otherwise transact with us, if
third parties perceive a higher than acceptable risk to commercialization of our products or future products. It
is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If we fail to adequately
protect our product candidates, others could compete against us more directly. Our
commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current
and future product candidates, the processes used to manufacture them and the methods for using them, as well as successfully
defending these patents against third-party challenges. The
patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions
for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceutical
patents has emerged to date in the United States or in foreign jurisdictions outside of the United States. Changes in either the
patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual
property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that may be issued from the
applications we currently or may in the future own or license from third parties. Further, if any patents we obtain or license
are deemed invalid and unenforceable, our ability to commercialize or license our technology could be adversely affected. The
degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may
not adequately protect our rights or permit us to gain or keep our competitive advantage. For example: We
cannot be certain that any future patents will issue with claims that cover our product candidates. Our ability to stop third
parties from making, using, selling, offering to sell, or importing our product candidates is dependent upon the extent to which
we have rights under valid and enforceable patents or trade secrets that cover these activities. If
we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology
and products could be significantly diminished. We
also rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate
or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees,
consultants, outside scientific collaborators, sponsored researchers, and other advisors to protect our trade secrets and other
proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide
an adequate remedy in the event of unauthorized disclosure of confidential information. Furthermore,
any license agreements we enter in the future may require us to notify, and in some cases license back to the licensor, certain
additional proprietary information, or intellectual property that we developed using the rights licensed to us under these agreements.
Any such licenses back to the licensor could allow our licensors to use that proprietary information or intellectual property
in a manner that could harm our business. In addition, others may independently discover our trade secrets and proprietary information.
For example, the FDA, as part of its transparency initiative, is currently considering whether to make additional information
publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information,
and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and
time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain
or maintain trade secret protection could adversely affect our competitive business position. We
may incur substantial costs because of litigation or other proceedings relating to patents and other intellectual property rights. If
we choose to commence a proceeding or litigation to prevent another party from infringing our patents, that party will have the
right to ask the examiner or court to rule that our patents are invalid or should not be enforced against them. There is a risk
that the examiner or court will decide that our patents are not valid and that we do not have the right to stop the other party
from using the related inventions. There is also the risk that, even if the validity of our patents is upheld, the examiner or
court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to
such patents. In addition, the U.S. Supreme Court has recently modified some tests used by the U.S. Patent and Trademark Office,
or USPTO, in granting patents over the past 20 years, which may decrease the likelihood that we will be able to obtain patents
and increase the likelihood of challenge to any patents we obtain or license. Any
proceedings or litigation to enforce our intellectual property rights or defend ourselves against claims of infringement of third-party
intellectual property rights could be costly and divert the attention of managerial and scientific personnel, regardless of whether
such litigation is ultimately resolved in our favor. We may not have sufficient resources to bring these actions to a successful
conclusion. Some of our competitors who may assert infringement may be able to sustain the costs of complex patent litigation
more effectively than we can because they are better capitalized and have more resources than us. Moreover, any uncertainties
resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise
the funds necessary to continue our operations. If
we are unable to successfully defend against claims that we have infringed the intellectual property rights of others, we may
be prevented from using certain intellectual property and may be liable for damages, which in turn could materially adversely
affect our business, financial condition, or results of operations. Alternatively, we could be compelled to seek licenses from
one or more third parties who could be direct or indirect competitors and who might not make licenses available on terms that
we find commercially reasonable or at all. Risks
Related to International Operations We
may be subject to extensive regulations outside the United States and may not obtain marketing approvals for products in Europe
and other jurisdictions. In
addition to regulations in the United States, should we or our collaborators pursue marketing approvals for Generx and our other
product candidates internationally, we and our collaborators will be subject to a variety of regulations in other jurisdictions
governing, among other things, clinical trials and any commercial sales and distribution of our products. Whether or not we, or
our collaborators, obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign
countries prior to the commencement of clinical trials or marketing of the product in those countries. The requirements and process
governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. We
expect to pursue marketing approvals for Generx and our other product candidates in Europe and other jurisdictions outside the
United States with collaborative partners. The time and process required to obtain regulatory approvals and reimbursement in Europe
and other jurisdictions may be different from those in the United States, and regulatory approval in one jurisdiction does not
ensure approvals in any other jurisdiction; however, negative regulatory decisions in any jurisdiction may have a negative impact
on the regulatory process in other jurisdictions. Following
a national referendum and enactment of legislation by the government of the United Kingdom, the United Kingdom withdrew from the
European Union, or Brexit, on January 31, 2020, and entered into a transition period during which it will continue its ongoing
and complex negotiations with the European Union relating to the future trading relationship between the parties. Significant
political and economic uncertainty remains about whether the terms of the relationship will differ materially from the terms before
withdrawal, as well as about the possibility that a so-called “no deal” separation will occur if negotiations are
not completed by the end of the transition period. Any delay in obtaining, or an inability to obtain, any marketing approvals,
as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the United Kingdom and/or
the European Union and restrict our ability to generate revenue and achieve and sustain profitability. If any of these outcomes
occur, we may be forced to restrict or delay efforts to seek regulatory approval in the United Kingdom and/or European Union for
our product candidates, which could significantly and materially harm our business. We
have entered into agreements with third parties to market our Generx product candidate in certain territories if approved by relevant
regulatory authorities, but there can be no assurance that the efforts of such third parties will meet our expectations or result
in any significant product sales. We
have entered into license agreements with Pineworld Capital Ltd, and Shanxi for the right to manufacture and sell Generx in greater
China and the CIS. The licenses are effective upon FDA approval to market Generx in the United States. Our licenses to
Pineworld Capital Ltd, and Shanxi are exclusive, and we do not have a right to separately manufacture, use or sell our Generx
product candidate into those territories. Consequently, we are dependent on the resources, efforts, and success of our licensees
to successfully develop a market for Generx in those territories. We do not control the operations of our licensees and have limited
rights to terminate the licenses under the terms of our agreements. We cannot be certain that our licensees will successfully
generate any significant product sales, or that the royalties that we ultimately receive from these arrangements will meet our
expectations.
Collaborations
with Third Parties outside the United States presents additional risks. Conducting
clinical trials in foreign countries, as we may do for our current and future product candidates, presents additional risks that
may delay completion of our clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere
to the clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative
burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries. To
the extent we agree to work exclusively with one collaborator in each area, our opportunities to collaborate with other entities
could be curtailed. Lengthy negotiations with potential new collaborators may lead to delays in the research, development, or
commercialization of product candidates. The decision by our collaborators to pursue alternative technologies or the failure of
our collaborators to develop or successfully commercialize any product candidate to which they have obtained rights from us could
materially harm our business, financial condition, and results of operations. To
the extent that we enter markets outside the United States, our business will be subject to political, economic, legal, and social
risks in those markets, which could adversely affect our business. There
are significant regulatory and legal barriers in markets outside the United States that we must overcome to the extent we enter
or attempt to enter markets in countries other than the United States. We will be subject to the burden of complying with a wide
variety of national and local laws, including multiple and possibly overlapping and conflicting laws. We also may experience difficulties
adapting to new cultures, business customs and legal systems. Any sales and operations outside the United States would be subject
to political, economic, and social uncertainties including, among others: Any
changes related to these and other factors could adversely affect any business operations that we conduct outside the United States. Risks
Related to Financial Position, Need for Additional Capital, and Worldwide Environment We
have incurred losses since inception and anticipate that we will continue to incur significant net losses for the foreseeable
future and may never achieve or maintain profitability. We
have sustained operating losses since our inception and will likely continue to sustain losses as we seek to develop our products
and product candidates. We expect these losses to be substantial because of the significant amounts we expect to spend on development
activities and clinical trials for our product candidates. We expect our net losses from operations to continue for at least the
next few years. Whether
we will generate additional revenues and become profitable will depend on our ability, alone or with potential collaborators,
to efficiently and successfully complete the development of our product candidates, successfully complete pre-clinical and clinical
tests, obtain necessary regulatory approvals, and manufacture and market our products. There can be no assurance that any such
events will occur or that we will ever become profitable. Even if we do achieve profitability, we cannot predict the level of
such profitability. If we sustain losses over an extended period of time, we may be unable to continue our business. We
will need substantial additional funding to develop our Generx product candidate, and if we are unable to raise capital when needed,
we could be forced to delay, reduce, or eliminate our product development programs or commercialization efforts. We
expect that our current cash will support our administrative operations only into 2021. Our expenses will increase over the next
several years as we continue to develop and conduct clinical trials with respect to our Generx or other product candidates, seek
regulatory approvals, and initiate commercialization efforts. Accordingly, we will be required to obtain further funding through
public or private equity offerings, debt financings, collaborations and licensing arrangements or other sources. We do not have
any arrangements for future financing in place currently. If we are unable to obtain such funds when needed, we may have to delay,
scale back or terminate our product development or our business. To
the extent we raise additional capital through the sale of equity securities, the ownership position of existing stockholders
could be substantially diluted. Anti-dilution adjustments to our Series A Convertible Preferred Stock and Series B Convertible
Preferred Stock could cause further dilution. If additional funds are raised through the issuance of preferred stock or debt securities,
these securities are likely to have rights, preferences and privileges senior to our common stock and may involve significant
fees, interest expense, restrictive covenants, and the granting of security interests in our assets. Future
sales of securities could result in additional dilution of the percentage ownership of our stockholders and could cause the share
price for our Common Stock to fall. We
expect that significant additional capital will be needed in the future to continue our planned operations, including conducting
clinical trials, hiring new personnel, commercializing our products, and continuing activities as an operating public company.
We expect to raise additional capital through the sale of debt or equity securities, but we do not have any firm arrangements
for capital in place currently. To the extent we raise additional capital by issuing equity securities, our stockholders may experience
substantial dilution. We may sell common stock, preferred stock or other convertible securities or other equity securities in
one or more transactions at prices and in a manner, we determine from time to time. If we sell common stock, convertible securities,
or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may
also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders. We
do not know how much additional financing will be necessary to finance our continued operations, which creates additional risk
that financing will not be available to us when needed, or that the terms may not be favorable or may result in additional dilution
to our current stockholders. Our
estimate as to how long we expect our existing cash to be able to continue to fund our operations and the costs required to move
our Generx product candidate to commercialization are based on assumptions that may prove to be wrong, and we could use our available
capital resources sooner than we currently expect. Further, changing circumstances, some of which may be beyond our control, could
cause us to consume capital significantly faster than we currently anticipate, and we may need to seek additional funds sooner
than planned. Our future funding requirements, both short-term and long-term, will depend on many factors, including: Because
of the numerous risks and uncertainties associated with product development, we are unable to accurately predict the timing or
amount of expenses or when, or if, we will obtain marketing approval to commercialize any of our product candidates. If we are
required by the U.S. Food and Drug Administration, or FDA, or other regulatory authorities such as the EMA, to perform studies
and trials in addition to those currently expected, or if there are any delays in the development, or in the completion of any
planned or future preclinical studies or clinical trials of our current or future product candidates, our expenses could increase,
and profitability could be further delayed. As
a result, we cannot predict with certainty the amount of capital that we will need to raise to finance our continued operations.
If we encounter unexpected delays or expenses or setback in our product development efforts, we may be compelled to seek additional
financing, which may not be available on terms that are favorable to our investors at that time. Our
recurring losses from operations raise substantial doubt regarding our ability to continue as a going concern. Our
consolidated financial statements for the years ending December 31, 2019, 2018 and 2017 were prepared under the assumption that
we will continue as a going concern for the next twelve months from the issuance date of these financial statements. Due to our
recurring losses from operations from our inception and our limited cash resources, we concluded that there is substantial doubt
in our ability to continue as a going concern within one year after the financial statements are issued without additional capital
becoming available. Our independent registered public accounting firm has issued an audit opinion that included an explanatory
paragraph referring to our projected future losses along with recurring losses from operations and expressing substantial doubt
in our ability to continue as a going concern without additional capital becoming available. Our ability to continue as a going
concern is dependent upon our ability to obtain additional equity or debt financing, attain further operating efficiencies, reduce
expenditures, and, ultimately, to generate revenue. The financial statements do not include any adjustments that might result
from the outcome of this uncertainty. We
are currently dependent on the services of a few key employees and need to increase the size of our organization. As
of December 31, 2019, we employed a total of four full-time employees. We will need to expand our managerial, operational, technical,
scientific, financial, and other resources to manage our operations and clinical trials, continue our research and development
activities, and commercialize our product candidate. Our management and scientific personnel, systems, and facilities currently
in place may not be adequate to support our future growth, and the loss of one or more of our executive officers or key employees
or an inability to attract and retain highly skilled employees could adversely affect our business. We will need to attract and
retain enough talented employees to: Competition
for qualified personnel is intense among companies, academic institutions, and other organizations. The pool of qualified personnel
with experience working with the pharma market is limited overall. In addition, many of the companies with which we compete for
experienced personnel have greater resources than we have. If we are unable to attract and retain key personnel, it may negatively
affect our ability to successfully develop, test, commercialize and market our products and product candidates. If we fail to
secure sufficient qualitied and talented personnel our development efforts may be delayed, become more costly or more susceptible
to failure. We
may have material weaknesses in our internal control over financial reporting which may result in misstatements in our financial
statements or erode investor confidence. We
have had limited financial resources and have historically had material weaknesses in our internal control over financial reporting,
as described elsewhere in this report. We have applied a portion of the funds secured from the Nostrum financing to enhance and
strengthen our internal controls and financial reporting. If we fail to completely mitigate those material weaknesses or significant
deficiencies in our internal controls continue or occur in the future, we may fail to meet our future reporting obligations on
a timely basis, or our financial statements could contain errors or misstatements. If such errors were sufficiently material,
we would be required to restate prior period financial results, which may subject us to class action litigation. Any
failure to address the ineffectiveness of our internal controls could also adversely affect the periodic management evaluations
of the effectiveness of our internal controls over financial reporting and our disclosure controls and procedures that are required
to be included in our annual report on Form 10-K. Continued reporting of internal control deficiencies could also cause investors
to lose confidence in our reported financial information, which could adversely impact demand for stock and stock price. We
now plan to file our quarterly reports on Form 10-Q for the quarters ended March 31, 2020, June 30, 2020 and September 30, 2020
and become current with our Section 13(a) filing obligations under the Securities Exchange Act of 1934. If remedial measures become
required or if material weaknesses or significant deficiencies in our internal controls continue or occur in the future, any of
the following may occur: Any
failure to address the ineffectiveness of our disclosure controls and procedures could also adversely affect the periodic management
evaluations of the effectiveness of our internal controls over financial reporting and our disclosure controls and procedures
that are required to be included in our annual report on Form 10-K. Internal control deficiencies and ineffective disclosure controls
and procedures could also cause investors to lose confidence in our reported financial information. The future measures we plan
to take may not remediate the ineffectiveness of our disclosure controls and procedures, and material weaknesses and restatements
of financial results may arise in the future due to a failure to implement and maintain adequate internal control over financial
reporting and adequate disclosure controls and procedures. In addition, even if we are successful in strengthening our controls
and procedures, in the future those controls, and procedures may not be adequate to prevent or identify irregularities or errors
or to facilitate the fair presentation of our consolidated financial statements. We
are not current with our reporting requirements under Section 13(a) of the Securities Exchange Act of 1934. We
suspended our public reporting beginning in 2017 due to financial hardship. Following the filing of our Annual Report on Form
10-K for the 2019, 2018 and 2017 fiscal years, which is covered by this filing, we plan to submit our Quarterly Reports on Form
10-Q for the quarterly periods ended March 31, 2020, June 30, 2020, and September 30, 2020, and become current with our reporting
obligations, and thereafter resume a timely filing schedule with respect to our future SEC reports. We expect to continue to face
many of the risks and challenges related to the matters that led to the delay in the filing of our Annual Report on Form 10-K
for the years ending December 31, 2017 and 2018, and 2019, including the following: If
one or more of the foregoing risks or challenges persist, our business, operations and financial condition are likely to be materially
and adversely affected. Impact
of Coronavirus Outbreak On
January 30, 2020, the World Health Organization (“WHO”) announced a global health emergency because of a new strain
of coronavirus originating in Wuhan, China (the “COVID-19 outbreak”) and the risks to the international community
as the virus spreads globally beyond its point of origin. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic,
based on the rapid increase in exposure globally and since then authorities throughout the world have implemented measures to
contain or mitigate the spread of the virus, including physical distancing, travel bans and restrictions, closure of non-essential
businesses, quarantines, work-from-home directives, and shelter-in-place orders. These measures have caused, and are continuing
to cause, business slowdowns or shutdowns in affected areas, both regionally and worldwide, which have impacted our business and
results of operations. The
full impact of the COVID-19 outbreak continues to evolve as of the date of this report. As such, it is uncertain as to the full
magnitude that the pandemic will have on the Company’s financial condition, liquidity, and future results of operations.
Management is actively monitoring the impact of the global situation on its financial condition, liquidity, operations, suppliers,
industry, and workforce. Given the daily evolution of the COVID-19 outbreak and the global responses to curb its spread, the Company
is not able to estimate the effects of the COVID-19 outbreak on its results of operations, financial condition, or liquidity for
fiscal year 2020 and 2021. Risks
Related to Our Capital Structure and Owning our Common Stock Our
outstanding shares of Preferred Stock and warrants to purchase Common Stock far exceed the number of shares of our Common Stock
outstanding and their conversion or exercise will result in substantial dilution to holders of our Common Stock. As
of March 31, 2021, we We
may consider affecting a reverse stock split or other share recapitalization transaction, which could impact the value of our
Common Stock. The
total number of shares of our Common Stock, on a fully diluted basis, nearly exceeds our authorized capital. In addition, we would
like to increase the per share price of our outstanding Common Stock to a Nostrum’s
control of approximately 75.2% of our
voting securities gives them control over any action requiring stockholder approval and may discourage some investors from investing.
Nostrum
through its ownership of our Series A Convertible Preferred Stock and Series B Convertible Preferred Stock controls approximately
75.2% of the voting interests our company. Nostrum will control the outcome of matters submitted to our stockholders for
approval, including the election of directors and any merger, consolidation, or sale of all or substantially all our assets. In
addition, Nostrum will exercise significant control over the management and affairs of our company. This concentration of ownership
might harm the market price of our Common Stock if: Our
Common Stock is not listed on a national exchange which may diminish the market interest, liquidity, and price for our Common
Stock. Our
common stock currently is listed only on the OTC Pink Sheets. We hope to have our Common Stock re-established on the OTC QB once
our SEC filing delinquencies are rectified. OTC QB is a reporting service and not a securities exchange. It is our intent to secure
a listing on the Nasdaq Capital Market or another National Exchange, but we do not currently meet the listing criteria and we
may never qualify for trading on a national exchange. Many
institutional investors are prohibited from investing in stock unless they are listed on a national exchange. Also index funds
are generally restricted to exchange listed securities. Accordingly, stock listed on the over-the counter market is less likely
to secure general market interest, including analyst and research coverage. Stocks that trade on the over-the-counter market may
experience lower trading volumes, higher spreads between bid and ask pricing, increased volatility, and lower prices generally
that those traded on a national exchange. The inability to list our Common Stock on a national exchange may negatively impact
the volume of trading and market price for our common stock. We
are subject to SEC rules concerning the regulation of “penny stocks” which may reduce investor demand and market prices
for our Common Stock. Our
Common Stock is currently a “penny stock” under applicable SEC rules. While we have that designation, broker-dealers
trading in our common stock must make a special suitability determination for the purchaser and receive the purchaser’s
written agreement to the transaction prior to the sale. This requirement may impair the ability of broker-dealers to sell our
Common Stock and the ability of interested purchasers to acquire shares. In addition to additional SEC regulation, penny stocks
are generally perceived as more susceptible to trading manipulation schemes such as (a) control of the market by one or a few
broker dealers, (b) manipulation of pricing through wash sale transactions, (c) so-called “boiler room” practices
involving high pressure sales tactics, (d) excessive and undisclosed bid-ask differentials and mark-ups by selling broker-dealers.
Consequently, many institutional investors will not invest in stock that are classified as penny stocks. These circumstances may
reduce the demand for our Common Stock and could result in reduced liquidity or lower market prices for our Common Stock. To
raise capital to fund the development of our Generx product candidate, we have sold shares in our Angionetics subsidiary. In
2016 we sold a 15% interest in our Angionetics, Inc. subsidiary to Pineworld Capital Limited. Our management did this because
it believed that it could raise capital at a better valuation, and with less dilution to existing stockholders, than if it were
to sell shares of The
price of our Common Stock
The
market price for our Common Stock may be subject to greater volatility than other stock as a result of:
We
do not anticipate generating cash from operations for several years while we continue development and qualification of our product
candidates. For the foreseeable future, we intend to retain any future earnings for the development, We
are a “smaller reporting company” and can avail ourselves of reduced disclosure requirements applicable to small reporting
companies, which could We
are a smaller reporting company, and we will remain a smaller reporting company until the fiscal year following the determination
that our voting and non-voting common stock held by non-affiliates is more than $250 million measured on the last business day
of our second fiscal quarter, or our annual revenues are more than $100 million during the most recently completed fiscal year
and our voting and non-voting common stock held by non-affiliates is more than $700 million measured on the last business day
of our second fiscal quarter. Smaller reporting companies can provide simplified executive compensation disclosure, are exempt
from the auditor attestation requirements of Section 404, and have certain other reduced disclosure obligations, including, among
other things, being required to provide only two years of audited financial statements and not being required to provide selected
financial data, supplemental financial information, or risk factors. We have elected to take advantage of certain of the reduced
reporting obligations, which may render our common stock less attractive to some investors. Our
charter and Delaware law have anti-takeover effects that could discourage, delay, or prevent a change in control, which may discourage
third party offers to acquire our Company. Our
company could be difficult to acquire due to anti-takeover provisions in our charter and Delaware law. In
addition, we are subject to the anti- takeover provisions of Section 203 of the Delaware General Corporation Law. Subject to specified
exceptions, this section provides that a corporation may not engage in any business combination with any interested stockholder
during the three-year period following the time that such stockholder becomes an interested stockholder. This provision could
have the effect of delaying or preventing a change of control of our company. The foregoing factors could reduce the price that
investors or an acquirer might be willing to pay in the future for shares of our common stock.
●
the
size and nature of the patient population;
●
the
proximity of patients to clinical sites; -19-
●
the
eligibility criteria for the clinical trial;
●
the
design of the clinical trial;
●
the
size of the patient population required for analysis of the trial’s primary endpoints;
●
our
ability to recruit clinical trial investigators with the appropriate competencies and experience;
●
our
ability to obtain and maintain patient consents;
●
the
risk that patients enrolled in clinical trials will drop out of the
●
competing
clinical trials and -20- -21-
●
timing
of market approval and commercial launch of Generx and our other product candidates;
●
the
clinical indication(s) for which Generx and our other product candidates are approved;
●
product
label and package insert requirements;
●
physician
and patient perception of the safety and efficacy of our products; -22-
●
strength
of sales, marketing, and distribution support;
●
product
pricing relative to alternative treatments;
●
future
changes in health care laws, regulations, and medical policies; and
●
availability
of reimbursement codes and coverage in select jurisdictions, and future changes to reimbursement policies of government and
third-party payors.
●
fines,
restitution, or disgorgement of profits or revenues;
●
restrictions
on the labeling or marketing of products;
●
restrictions
on product manufacturing, distribution or use;
●
requirements
to conduct post-marketing studies or clinical trials;
●
warning
letters or untitled letters;
●
refusal
to approve pending applications or supplements to approved applications that we submit;
●
recall
of products;
●
or
withdrawal of products from the market; or
●
injunctions
or the imposition of civil or criminal penalties. -23-
●
sourcing
and producing raw materials;
●
transferring
technology from chemistry and development activities to production activities;
●
validating
initial production designs;
●
scaling
manufacturing techniques:
●
improving
costs and yields;
●
establishing
and maintaining quality controls and stability requirements;
●
eliminating
contaminations and operator errors; and
●
maintaining
compliance with regulatory requirements. -24-
●
labor
disputes, resource constraints, shipment delays, or inventory shortages;
●
product
loss due to contamination, equipment failure or improper installation or operation of equipment, or vendor or operator error;
●
reduced
production yields, product defects, and other supply disruptions due to deviations, even minor, from normal manufacturing
and distribution processes;
●
microbial,
viral, or other contaminations in our product candidate or in the manufacturing facilities in which our product candidate
is made, which may result in the closure of such manufacturing facilities for an extended period of time to allow for the
investigation and remediation of the contamination;
●
lawsuits
related to our manufacturing techniques, equipment used during manufacturing, or composition of matter;
●
unstable
political environments, acts of terrorism, war, natural disasters, and other natural and man-made disasters. -25- -26-
●
others
may be able to make compounds that are similar to our product candidates, but that are not covered by the claims of our patents;
●
we
might not have been the first to make the inventions covered by our pending patent applications;
●
we
might not have been the first to file patent applications for these inventions;
●
our
patent applications may not result in issued patents;
●
the
claims of our issued patents or patent applications when issued may not cover our products or product candidates;
●
any
patents that we obtain may not provide us with any competitive advantages;
●
any
granted patents may be held invalid or unenforceable as a result of legal challenges by third parties;
●
the
patents of others may have an adverse effect on our business; and
●
there
may be significant pressure on the United States government and other international governmental bodies to limit the scope
of patent protection both inside and outside the United States for treatments that prove successful as a matter of public
policy regarding worldwide health concerns. -27- -28-
●
changes
and limits in import and export controls;
●
increases
in custom duties and tariffs;
●
changes
in currency exchange rates;
●
economic
and political instability;
●
changes
in government regulations and laws;
●
absence
in some jurisdictions of effective laws to protect our intellectual property rights; and
●
currency
transfer and other restrictions and regulations that may limit our ability to sell certain products or repatriate profits
to the United States. -29-
●
the
scope, progress, timing, costs, and results of clinical trials of Generx and our other product candidates;
●
the
costs, timing, and outcome of seeking regulatory approvals;
●
our
ability to enter into, and the terms and timing of, any collaboration arrangements
●
the
costs of commercialization activities for any of our product candidates that receive marketing approval;
●
our
overhead growth and associated costs;
●
revenue
received from commercial sales, if any, of our current and future product candidates;
●
changes
in regulatory policies or laws that may affect our operations; or
●
competing
technological and market developments. -30-
●
manage
our clinical trials effectively, including our planned clinical trials of Generx;
●
manage
our internal development efforts;
●
establish
and manage contract relationships with third parties; and
●
improve
our operational, financial and management controls and reporting systems. -31-
●
we
will continue to fail to meet our future reporting obligations on a timely basis;
●
our
consolidated financial statements may contain material misstatements;
●
we
could be required to restate our prior period financial results;
●
our
operating results may be harmed;
●
we
may be subject to class action litigation; and
●
we
may be unable to list our Common Stock on a National Exchange.
●
failure
to timely file our SEC reports and make our current financial information available, has placed, and will continue to place,
downward pressure on our stock price;
●
further
delay in the filing of our SEC reports will delay our ability to seek the relisting of our common stock on a national securities
exchange, and as a result, may continue to reduce the liquidity of our common stock;
●
litigation
and claims as well as regulatory examinations, investigations, proceedings, and orders arising out of our failure to file
SEC reports on a timely basis will continue to divert management attention and resources from the operation of our business;
●
we
may not be able to recapture lost business or business opportunities due to ongoing reputational harm; and
●
negative
reports or actions on our commercial credit ratings would increase our costs of, or reduce our access to, future commercial
credit arrangements and limit our ability to refinance existing indebtedness. -32-
●
Our
stockholders generally perceive that Nostrum’s goals as a shareholder differ from their own;
●
Activist
investors are dissuaded from investing because they cannot secure meaningful control; and -33-
●
Potential
acquirors would be discouraged from making a tender offer or otherwise attempting to gain control of the company; or
●
Nostrum
determines to a significant portion of its holdings.
●
the
limited size of our public float;
●
our
stock trading on the over the counter market;
●
our
dependence on a single or limited number of products candidates;
●
the
binary nature of the drug development and approval process; and
●
our
current capital structure.
our Common Stock is risky, and you should invest in our common stock only if you can withstand a significant loss and wide
fluctuations in the market value of your investment. Moreover, substantial volatility in our trading price would increase the
potential for us to be subject to shareholder lawsuits that, even if unsuccessful, could be costly to defend and a distraction
management time and resources away from our core operations.
ITEM
1B.
UNRESOLVED
STAFF COMMENTS
| MINE SAFETY DISCLOSURES |
Not applicable.
| -35- |
MARKET
FOR OUR COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our
common stock currently trades on the OTC
As
of December 21,
Dividends
We
have never declared or paid any cash dividends on our common stock and we do not intend to declare or pay a dividend in the foreseeable
future
Recent Sales of Unregistered Securities
We
did not issue any securities in unregistered transactions during the years ended December 31,
Repurchases of Equity Securities
We
did not repurchase any of our
Equity Compensation Plan Information
We
do not currently have an equity incentive plan
The
following table summarizes equity compensation plans approved by stockholders and equity compensation plans that were not approved
by stockholders as of December 31,
| Plan Category | (a) Number of securities to be issued upon exercise of outstanding options, warrants and rights | (b) Weighted average exercise price of outstanding options, warrants and rights | (c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||||
| Equity compensation plans approved by stockholders | — | $ | — | — | ||||||||
| Equity compensation plans not approved by stockholders | 12,111,333 | $ | 0.71 | — | ||||||||
| Total | 12,111,333 | $ | 0.71 | — | ||||||||
As
a smaller reporting company, we are not required to provide the information required by this item.
| -36- |
| MANAGEMENT |
The
following discussion and analysis
We
are a clinical stage biotechnology company focused on pre-clinical, clinical
We operated throughout the period covered by this report, with severely limited financial resources. During 2015 and 2016, prior to the period covered in this report, we took significant actions to reduce our operating expenses, including headcount reductions, downsizing offices, and suspending some operations while we sought capital to continue our business operations. In 2016 we contributed our assets related to our Generx product candidate into our Angionetics, Inc. subsidiary. We then sold a 15% preferred equity ownership interest in Angionetics, Inc. to Huapont in exchange for $3.0 million. After the filing of our quarterly report for the period ended March 31, 2017, we suspended filing our periodic reports with the SEC because we lacked the financial resources to continue the financial statement review and audits. This report covers our results of operations for the years ended December 31, 2019, 2018 and 2017.
Our
current business is focused exclusively on the development of Generx, a gene therapy product candidate targeted for men and women
with advanced ischemic heart disease and refractory angina. We have received FDA
Significant Developments
During the period covered by this report we entered into the following significant transactions:
| ● | In October 2017 we entered into an agreement with Landmark to assist us in our efforts to sell our Excellagen product and assist with the strategic partnering for the development of Generx. In lieu of an initial cash engagement fee of $50,000, we assigned our residual investment in LifeAgain along with a minority equity investment in Healthy Brands to Landmark, effectively exiting those businesses. We recorded this initial engagement fee as a consulting cost and the transfer of the assets, which had a net book value of zero, as a gain on transfer of assets and licenses in other income in the statement of operations. In connection with this agreement, and in exchange for business advice and marketing of the business for the purposes of raising financing, we issued Landmark a ten-year warrant to purchase up to 2.0 million shares of our Common Stock at a price of $0.25 per share. The fair value of the warrants was determined, using the Black-Scholes-Merton model, to be $230,000 and was recorded in the statement of operations as consulting services in selling, general and administrative expenses the period in which the services were rendered. | |
| ● | On November 14, 2017, we issued 700,000 warrants to a consultant for general business and scientific consulting services. The fair value of these warrants was determined to be $79,223 and was recorded in the statement of operations as consulting services in selling, general and administrative expenses in the period in which the services were rendered. | |
| ● | In August 2018, we sold our Excellagen® product to Olaregen for aggregate consideration of up to $4.0 million. At closing, we received a cash payment of $650,000, plus royalty payments of 10% of all worldwide sales of Excellagen outside of China, the Russian Federation, and the CIS, up to an additional $3,350,000. We recognized the gain on sale of Excellagen® in the amount of $650,000 during our third quarter ended September 30, 2018. The remaining $3,350,000 in additional consideration will be recognized as a gain in the periods that Olaregen reports sales that are subject to royalty and collection is reasonably assured. To date no royalty payments have been received. |
| -37- |
| ● | During 2019, we took a number of measures to restructure our accounts payable to third party vendors, including negotiated settlements with vendors that resulted in forgiveness of a portion of the accounts payable. For the year ended December 31, 2019, we recognized in our Statement of Operations $1,659,917 as a gain on re-negotiation of vendor payables. The total gain on re-negotiation includes $397,449 in restructured amounts that become due and payable when and if the Company receives FDA approval or when the Company commercializes. For amounts that are payable contingent upon FDA approval or commercialization, the Company has recognized included the amount in the gain on forgiveness and disclosed the contingent payable since the timing and ultimate payment is not determinable. As of December 31, 2019, we had outstanding trade payable and accrued liabilities of $3,763,816. |
Subsequent Events
The following significant events took place after the period covered by this report:
| ● | On September 10, 2019, December 30, 2019, and April 30, 2020, we issued Nostrum a promissory note in exchange for cash of $120,000 on September and December 2019 and $25,000 on April 30, 2020. These bear interest at 6% per annum and mature 24 months from the date of issuance. The cash funding related to the December 30, 2019 promissory note was not received by the Company until January 2020, so the Company recorded the note payable in the consolidated balance sheet in January 2020, upon receipt of the cash from Nostrum. | |
| ● | As of December 31,
2019, we had an outstanding balance in accrued but unpaid salaries and benefits for current and former employees totaling
$2,866,717. In January 2020, all affected current and former employees agreed to defer their compensation, less
applicable tax withholdings, upon the earliest to occur of (a) the FDA’s approval of Generx for marketing and sale in
the U.S.; (b) the EMA approval of Generx for marketing and sale in the European Union and the United Kingdom; (c) the
sale of Generx to an independent third party for an aggregate value equal to or greater than $35,000,000; (d) our entry into
a strategic partnership that would facilitate a capital contribution equal to or greater than $35,000,000 for the purpose
of supporting the clinical and commercial development | |
| ● | On April 10, 2020, we entered into the Ratification Agreement with Shanxi. In connection with the Ratification Agreement, we terminated all prior agreements with Shanxi, cancelled a prepaid $600,000 equity subscription and entered into a mutual release of claims. | |
| ● | On April 10, 2020, our Angionetics, Inc. subsidiary entered into the Shanxi License Agreement, granting Shanxi certain license rights with respect to our Generx product candidate. The distribution and license rights commence only after we obtain U.S. FDA approval for marketing and sale of Generx in the United States. The license rights include (a) a non-exclusive right to manufacture Generx products in China, and (b) an exclusive right to market and sell Generx products in Singapore, Macau, Hong Kong, Taiwan, any other municipality other than mainland China where Chinese (Mandarin or Cantonese) is the common language, the Russian Federation, and the CIS (i.e., Armenia, Azerbaijan, Belarus, Kazakhstan, Kyrgyzstan, Moldova, Tajikistan, Turkmenistan, and Uzbekistan). The Shanxi License Agreement provided for payment of $600,000 upfront, which was paid by application of the prepaid equity subscription, and a royalty ranging from 5% up to 10% based on the level of annual net sales of the Generx product sold by Shanxi in the licensed territory. | |
| ● | On April 10, 2020,
our Activation Therapeutics, Inc. subsidiary entered into the Shanxi Assignment Agreement pursuant to which we transferred
all of our license rights to manufacture, use, market and sell Excellagen to Shanxi. We also assigned to Shanxi a Chinese
patent that we received on Excellagen. As a result, we no longer have an interest in Excellagen, other than the right to the
| |
| ● | In May 2020, we
entered into a Preferred Stock Purchase Agreement with |
| -38- |
| ● | The
Series B Convertible Preferred Stock financing resulted in a reset of the conversion price
| |
| ● | During 2020, we entered into additional settlement agreements with third party vendors resulting in additional gains on vendor payables of $68,032 on our accounts payable. | |
| ● | In March 2021, the Company entered into an agreement with FUJIFILM Diosynth Biotechnologies (“FDB”) to manufacture the Generx [Ad5FGF-4] angiogenic gene therapy product candidate for Phase 3 clinical evaluation for the treatment of refractory angina due to late-stage coronary artery disease. Manufacturing operations will be conducted at FDB’s facilities in College Station, Texas where FDB will perform technology transfer and process development activities for Phase 3 clinical and commercial-scale GMP manufacturing of Generx. |
Our
consolidated financial statements included
We
believe that the following accounting policies involve the most complex judgments concerning assumptions and estimates with the
greatest potential impact on our consolidated financial statements. Therefore, we consider these to be our critical accounting
policies and estimates. For further information on all of our significant accounting policies, see the notes to our consolidated
financial statements
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires us to make estimates and assumptions that affect the reported amounts of our assets, liabilities, revenues, and expenses, and that affect our recognition and disclosure of contingent assets and liabilities.
While our estimates are based on assumptions that we consider reasonable at the time they were made, actual results may differ from our estimates, perhaps significantly. If results differ materially from our estimates, we will adjust our financial statements prospectively as we become aware of the necessity for an adjustment.
We believe it is important for you to understand our most critical accounting policies. These are our policies that require us to make our most significant judgments and, as a result, could have the greatest impact on our future financial results.
Preferred Stock
The Company applies the accounting standards for distinguishing liabilities from equity when determining the classification and measurement of its preferred stock. Shares that are subject to mandatory redemption (if any) are classified as liability instruments and are measured at fair value. The Company classifies conditionally redeemable preferred shares, which includes preferred shares that feature redemption rights that are either within the control of the holder or subject to redemption upon the occurrence of uncertain events not solely within our control, as temporary equity. At all other times, preferred shares are classified as stockholders’ equity.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating losses and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income(loss) in the years in which those temporary differences are expected to be recovered or settled. Due to the Company’s history of losses, a full valuation allowance has been recognized against the deferred tax assets.
| -39- |
The Company has adopted the provisions of ASC 740-10, which clarifies the accounting for uncertain tax positions. ASC 740-10 requires that the Company recognize the impact of a tax position in its financial statements if the position is more likely than not to be sustained upon examination base on the technical merits of the position. For the year ended December 31, 2019, the Company had no material unrecognized tax benefits, and based on the information currently available, no significant changes in unrecognized tax benefits are expected in the next twelve months.
The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. For the years ended December 31, 2019, 2018 and 2017, the Company has not recorded any interest or penalties related to income tax matters. The Company does not foresee any material changes in unrecognized tax benefits within the next twelve months.
When tax returns are filed, there may be uncertainty about the merits of positions taken or the amount of the position that would be ultimately sustained. The benefit of a tax position is recognized in the financial statements in the period during which, based on all available evidence, management believes it is more likely than not that the position will be sustained upon examination, including the resolution of appeals or litigation processes, if any. Tax positions taken are not offset or aggregated with other positions.
Tax positions that meet the more likely than not recognition threshold is measured at the largest amount of tax benefit that is more than 50 percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefit associated with tax positions taken that exceed the amount measured as described above should be reflected as a liability for uncertain tax benefits in the accompanying balance sheet along with any associated interest and penalties that would be payable to the taxing authorities upon examination. The Company believes our tax positions are all more likely than not to be upheld upon examination. As such, the Company has not recorded a liability for uncertain tax benefits.
Warrants
Warrants issued to third parties in connection with consulting and other services do not trade in an active securities market, and as such, we estimate the fair value of these warrants using an option pricing model. Following the authoritative accounting guidance, warrants with variable exercise price features or with potential cash settlement outside of our control are accounted for as liabilities, with changes in the fair value included in operating expenses, otherwise warrants determined to be equity classified are fair valued at the date of issuance, with no change in the fair value recorded in subsequent periods. We estimated the fair value of the warrants using the Black Scholes option pricing model. The Black Scholes model requires that our management make certain estimates regarding the expected stock volatility, the risk–free interest rate, the warrant’s expected life, and the expected forfeiture rate, to derive an estimated fair market value.
Results of Operations
Fiscal
2019 Compared to Fiscal
The following tables sets forth our results of operations for the years ended December 31, 2019 and 2018, and the relative dollar and percentage change between the two years.
| Year
Ended December 31, | Change (2019 to 2018) | |||||||||||||||
| 2019 | 2018 | ($) | % | |||||||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | $ | 243,453 | $ | 255,394 | (11,941 | ) | (4.7 | )% | ||||||||
| Selling, general and administrative | 593,549 | 907,836 | (314,287 | ) | (34.6 | )% | ||||||||||
| Total Operating Expenses | 837,002 | 1,163,230 | (326,228 | ) | (28.0 | )% | ||||||||||
| Gain on sale of assets and technology | — | (650,000 | ) | 650,000 | 100 | % | ||||||||||
| Income (Loss) from Operations | (837,002 | ) | (513,230 | ) | 323,772 | 63.1 | % | |||||||||
| Other Income (Expense): | ||||||||||||||||
| Gain on forgiveness of account payables | 1,659,917 | — | 1,659,917 | 100 | % | |||||||||||
| Interest Expense | (43,787 | ) | (39,514 | ) | (4,273 | ) | 10.8 | % | ||||||||
| Total Other Income (Expense) | 1,616,130 | (39,514 | ) | 1,655,644 | (4,190.0 | )% | ||||||||||
| Net Income (Loss) | 779,128 | (552,744 | ) | 1,331,872 | 241.0 | % | ||||||||||
| Net (Loss) attributable to the non-controlling interest | (87,547 | ) | (117,863 | ) | 30,316 | (25.7 | )% | |||||||||
| Net Income (Loss) attributable to the controlling interest | 866,675 | (434,881 | ) | 1,301,556 | (299.3 | )% | ||||||||||
| -40- |
Research and development decreased in 2019 compared to 2018 by $11,941 or 4.7% due to a decrease in employee benefits.
Selling, general and administrative expenses decreased in 2019 by $314,287 or 34.6% compared to 2018 mainly due to a reduction in employee salary costs of $152,852 resulting from a headcount reduction of two employees on a permanent basis and one employee on a temporary basis during 2019. In addition, the Company incurred consulting costs of approximately $80,000 in relation to raising capital funds for the Company in 2018 compared with $nil in 2019, and an overall reduction in legal and regulatory professional fees of $61,851, in addition to a decrease in office supplies as the Company focused time and resources on raising capital resources and putting on hold regulatory filing matters therefore reducing the selling, general and administrative expenses in 2019 when compared to 2018. In addition, the depreciation expense in 2019 was lower due to Company property and equipment becoming fully depreciated.
During the year ended December 31, 2018, the company recognized a gain on sale of Excellagen® product to Olaregen in the amount of $650,000 which also represented the cash proceeds on the sale of the technological asset. The sale of Excellagen® was consistent with management restructuring of the company’s operations in order to focus efforts on development and sale of Generx.
Other expenses for the year ended December 31, 2019 included a gain on debt forgiveness in the amount of $1,659,917. The debt forgiveness is the result of settlement agreements reached with certain vendors as part of the pre-financing restructuring efforts of the Company. Of these amounts, $172,449 becomes due and payable upon FDA approval of Generx, and when total cumulative net sales of Generx reach $100 million, an additional amount totaling $225,000 will be due and payable. Interest expense increased in 2019 compared to 2018 by $4,273 primarily as result of an increase in the notes payable in the third quarter ended 2019 of approximately $120,000 bearing interest at 6% per annum on advances received from Nostrum.
Fiscal 2018 Compared to Fiscal 2017
The following tables sets forth our results of operations for the years ended December 31, 2018 and 2017, and the relative dollar and percentage change between the two years.
| Year
Ended December 31, | Change (2018 to 2017) | |||||||||||||||
| 2018 | 2017 | ($) | % | |||||||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | $ | 255,394 | $ | 344,976 | (89,582 | ) | (26.0 | )% | ||||||||
| Selling, general and administrative | 907,836 | 1,781,309 | (873,473 | ) | (49.0 | )% | ||||||||||
| Total Operating Expenses | 1,163,230 | 2,126,285 | (963,055 | ) | (45.3 | )% | ||||||||||
| Gain on sale of assets and technology | (650,000 | ) | (50,000 | ) | 600,000 | 1,200 | % | |||||||||
| Income (Loss) from Operations | (513,230 | ) | (2,076,285 | ) | 1,563,055 | (75.3 | )% | |||||||||
| Other Income (Expense): | ||||||||||||||||
| Interest Expense | (39,514 | ) | (20,219 | ) | (11,339 | ) | 56.1 | % | ||||||||
| Total Other Income (Expense) | (39,514 | ) | (20,219 | ) | (11,339 | ) | 56.1 | % | ||||||||
| Net Loss | (552,744 | ) | (2,096,504 | ) | (1,543,760 | ) | (73.6 | )% | ||||||||
| Net Loss attributable to the non-controlling interest | (117,863 | ) | (202,362 | ) | (84,499 | ) | (41.8 | )% | ||||||||
| Net Loss attributable to the controlling interest | (434,881 | ) | (1,894,142 | ) | (1,459,262 | ) | (77.0 | )% | ||||||||
Research and development decreased in 2018 compared to 2017 by $89,582 or 26 %. The decrease in spending is primarily due to the Company’s continued cash constrained position in 2018 resulting in management focusing Company resources on raising capital to provide the resources to advance the development and commercialization of Generx. The Company also discontinued further development of other product lines and focused actively on selling intellectual property developed not related to the Generx product line.
Selling,
general and administrative expenses
During
the year ended December 31, 2018,
| -41- |
Other expense for the year ended December 31, 2018 and 2017 also included interest expense related to interest on notes payable with unrelated parties. The total interest expense in 2018 was $31,558 compared with $20,219 in 2017, as a result of the note payable in the principal amount of $208,500, being outstanding for the full year in 2018 compared to an increase in the note through the 2017 year.
Fiscal 2017 Compared to Fiscal 2016
The following tables sets forth our results of operations for the years ended December 31, 2017 and 2016, and the relative dollar and percentage change between the two years.
| Year
Ended December 31, | Change (2017 to 2016) | |||||||||||||||
| 2017 | 2016 | ($) | % | |||||||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | $ | 344,976 | $ | 641,572 | (296,596 | ) | (46.2 | )% | ||||||||
| Selling, general and administrative | 1,781,309 | 2,199,412 | (418,103 | ) | (19.0 | )% | ||||||||||
| Total Operating Expenses | 2,126,285 | 2,840,984 | (714,699 | ) | (25.2 | )% | ||||||||||
| Gain on sale of assets and technology | (50,000 | ) | — | (50,000 | ) | 100.0 | % | |||||||||
| Income (Loss) from Operations | (2,076,285 | ) | (2,840,984 | ) | 764,699 | (26.9 | )% | |||||||||
| Other Income (Expense): | ||||||||||||||||
| Interest Expense | (20,219 | ) | (172,825 | ) | 152,606 | (88.3 | )% | |||||||||
| Total Other Income (Expense) | (20,219 | ) | (172,825 | ) | 152,606 | (88.3 | )% | |||||||||
| Net Loss | (2,096,504 | ) | (3,013,809 | ) | 917,305 | (30.4 | )% | |||||||||
| Net Loss attributable to the non-controlling interest | (202,362 | ) | (95,581 | ) | (106,781 | ) | 111.7 | % | ||||||||
| Net Loss attributable to the controlling interest | (1,894,142 | ) | (2,918,228 | ) | (1,024,086 | ) | (35.1 | )% | ||||||||
Research and development expense decreased in 2017 compared to 2016 by $296,596 0r 46.2% as a result of the Company reducing all discretionary expenses in 2017 in order to conserve cash and focus on raising capital to be used for the Company’s efforts to continue the development of their core technologies and due to a decrease in employee salary expenses as a result of a reduction in headcount.
Selling, general and administrative expenses decreased in 2017 compared to 2016 by $418,103 or 19% due to the Company reducing headcount and discretionary expenditures during 2017, including suspending SEC filings to reduce legal, accounting and filing costs.
The
Company recognized a gain on sale of assets in the amount of $50,000 in the year ended December 31, 2017. In October 2017,
the Company entered into an agreement with Landmark to assist with the Company’s efforts to sell the Excellagen product
and assist with the strategic partnering for the development of Generx. In lieu of an initial cash engagement fee of $50,000,
the Company assigned our residual investment in LifeAgain along with a minority equity investment in Healthy Brands to Landmark,
effectively exiting the development and commercialization of the product lines. The Company determined the fair value of the non-monetary
exchange to be $50,
Other
income/expense includes interest expense which decreased by $152,606 in 2017 compared with 2016. In 2016 $146,996 in interest
charges on unpaid license fees Liquidity
and Capital Resources The
following table summarizes our liquidity and working capital position on December 31, 2019, 2018 and 2017: Following
the period covered by this report: The
Company has In
2018, the Company sold Excellagen for cash proceeds of $650,000 resulting in cash being generated from investing activities. The
Company can also earn royalties on future sales on Excellagen, under the terms of the sales agreement. Net
cash provided by financing activities in 2019 compared to cash used by financing activities After
the period covered by this report, we secured the We
anticipate that negative cash flows from operations will continue for the foreseeable future.
We do not have any unused credit facilities. Our cash position, even after the Series B Convertible Preferred Stock financing
with Nostrum, will not be sufficient to sustain our operations for more than twelve months. We intend to secure additional working
capital to support our continued operations through sales of additional equity and debt securities Our
principal business objective is to
advance or Generx product candidate through Our
history of recurring losses and uncertainties as to whether our operations will become profitable raise substantial doubt about
our ability to continue as a going concern. Our consolidated financial statements do not include any adjustments related to the
recoverability of assets or classifications of liabilities that might be necessary should we be unable to continue as a going
concern. Off-Balance
Sheet Arrangements As
of December 31, 2019, we did not have any significant off-balance sheet debt, nor did we have any transactions, arrangements,
obligations (including contingent obligations) or other relationships with any unconsolidated entities or other persons that have
or are reasonably likely to have a material current or future effect on financial condition, changes in financial condition, results
of operations, liquidity, capital expenditures, capital resources, or significant components of revenue or expenses material to
investors. Recent
Accounting Pronouncements See
Note 2 to the Consolidated Financial Quarterly
Results of Operations For
the Three Months Ended March 31, 2019 compared to the Three Months Ended March 31, 2018 The
following tables sets forth our results of operations for the three-month period ended March 31, 2019 and 2018, and the relative
dollar and percentage change between the two periods. Change
(2019
to 2018) Selling,
general and administrative expenses decreased, for the three months ended March 31, in 2019 by $55,031 compared to 2018 mainly
due to a reduction in employees’ salary cost of approximately $24,000 resulting from two employees shifting from full-time
to part-time in 2019. In addition, the Other
expense increased $2,202 during the three-month period ended March 31, 2019 compared with March 31, 2018 primarily as a result
of increase in the interest rate and due to the compounded interest rate impact on the note payable in 2019. For
the Three Months and Six Months Ended June 30, 2019 compared to the Three Months and Six Months Ended June 30, 2018 The
following tables sets forth our results of operations for the three-month period and six-month ended June 30, 2019 and 2018, and
the relative dollar and percentage change between the two periods. Change (2019
to 2018) Change (2019
to 2018) Research
and development expense for the three-month and six-month periods ended June 30, 2019 and 2018 remained substantially the same
period over period. Selling,
general and administrative expenses decreased for the three-month period ended June 30, 2019 compared to the three-month period
ended June 30, 2018 by $87,156 or 32.6% primarily as a result of $80,000 in consulting costs in relation to raising capital funds
for the Company during the three-month period ended June 30, 2018 compared with $nil for the three-month period ended June 30,
2019. The Company’s employee salary costs also decreased by $18,958 as result of two employees transitioning from
full-time to part-time on a temporary basis, these decreased were offset by increases in miscellaneous office expenses. Selling,
general and administrative expenses decreased for the six-month period ended June 30, 2019 compared to the six-month period ended
June 30, 2018 by $142,187 or 28.3% primarily as a result of $105,000 in consulting costs in relation to raising capital funds
for the Company during the six-month period ended June 30, 2018 compared with $nil in the six-month period ended June 30, 2019.
In addition, salaries and benefits decreased by $44,374 resulting from two employees shifting from full-time to part-time
in 2019 on a temporary basis, offset by increases in miscellaneous office expenses. Other
income/expense increased for the three-month period ended June 30, 2019 compared to the three-month period ended June 30, 2018
by $1,406 or 15.5% primarily as a result of an increase in the interest rate on the notes payable effective May, 2018 and due
to the compounded interest impact on the note. Other
income/expense increased for the six-month period ended June 30, 2019 compared to the six-month period ended June 30, 2018 by
$3,608 or 21.2% primarily as a result of an increase in the interest rate on the outstanding notes payable effective May 2018
and due to the compounded interest impact on the note. For
the Three Months and Nine Months Ended September 30, 2019 compared to the Three Months and Nine Months Ended September 30, 2018 The
following tables sets forth our results of operations for the three-month period and Nine-month ended September 30, 2019 and 2018,
and the relative dollar and percentage change between the two periods. Change (2019
to 2018) Change (2019
to 2018) Research
and development expense decreased for the three-month period ended September 30, 2019 compared to three-month period ended September
30, 2018 by $4,999 or 7.5% primarily due to a decreased in employee salary and benefit expenses. Research
and development expense decreased for the nine-month period ended September 30, 2019 compared to nine-month period ended September
30, 2018 by 7,081 or 3.7% primarily due to a decrease in employee salary and benefit expenses and the company’s continued
cash constrained position resulting in management focusing company resources on raising capital to provide the resources to advance
the development and commercialization of Generx. Selling,
general and administrative expenses decreased for the three-months period ended September 30, 2019 compared to the three-month
period ended September 30, 2018 by $93,232 or 45.0% primarily as a result of decrease in employee wages and benefits by $56,875,
a decrease in consulting expenses by $27,000, and other discretionary expenditures. Selling,
general and administrative expenses decreased for the nine-month period ended September 30, 2019 compared to the nine-month period
ended September 30, 2018 by $235,419 or 33.2% mainly due to a reduction in employee salary and benefit expenses of $101,250 resulting
from a reduction in headcount and two employees moving from full-time to part-time on a temporary basis. In addition, the
company incurred consulting costs of approximately $130,000 in relation to raising capital funds in 2018 compared with $nil in
2019, and an overall reduction in other miscellaneous expenses as the company focused time and resources on raising capital. During
the three-month period and nine-month period ended September 2018, the company recognized a gain on sale of Excellagen technology
to Olaregen in the amount of $650,000, which also represented the cash proceeds on the sale. Other
income/expense increased for the three-month period ended September 30, 2019 compared to the three-month period ended September
30, 2018 by $37,363 or 298.6% primarily as a result of accounts payable forgiveness settlement agreements totaling $35,985, with
certain vendors as a part of the pre-financing restructuring efforts of the company. The increase in other income was offset by
an increase in interest expense for the nine-month period ended September 30, 2019 compared to the nine-month period ended September
30, 2018 by $1,651 primarily as a result of an increase in the notes payable of $120,000 received from Nostrum in September 2019,
which bears an interest at 6% per annum. For
the Three Months Ended March 31, 2018 compared to the Three Months Ended March 31, 2017 The
following tables sets forth our results of operations for the three-month period ended March 31, 2018 and 2017, and the relative
dollar and percentage change between the two periods.
Change
(2018
to 2017) Research
and development expense decreased for the three-month period ended March 31, 2018 compared to three-month period ended March 31,
2017 by $67,995 or 52.0% primarily due to a decrease in clinical trial expenses as a result of the Company focusing available
resources and time on raising capital for the ongoing development of the Generx product and restructuring the business to sell
all other assets and technology that the Company restructured as non-core products. Selling,
general and administrative expenses decreased for the three-month period ended March 31, 2018 compared to the three-month period
ended March 31, 2017 by $287,935 or 55.1% due to a decrease in employee salaries and benefits of approximately $72,280 as a result
of a decrease in headcount and reduced legal and other professional fees as a result of the Company’s decision to suspend
SEC filings beginning in 2017 of approximately $145,000 and a decrease in general office expenses, insurances costs and travel
costs of approximately $66,000. Other
income/expense increased for the three-month period ended March 31, 2018 compared to the three-month period ended March 31, 2017
by $5,970 or 305.1% primarily as a result of an increase in the notes payable during the third and fourth quarter of 2017, resulting
in an increase in interest expense. For
the Three Months and Six Months Ended June 30, 2018 compared to the Three Months and Six Months Ended June 30, 2017 The
following tables sets forth our results of operations for the three-month period and six-month ended June 30, 2018 and 2017, and
the relative dollar and percentage change between the two periods. Change
(2018
to 2017) Change (2018
to 2017) Research
and development expense for the three months ended June 30, 2018 compared with the three months ended June 30, 2017 remained consistent
and for the six months ended June 30, 2018 compared to the six months ended June 30, 2017 decreased $71,128 or 36.1%. The decrease
is primarily related to a decrease in clinical trial expenses of $63,028, decrease in employee salaries and benefits of approximately
$6,357 and a decrease in other miscellaneous expenses such as travel and supplies. Selling,
general and administrative expenses for the three months ended June 30, 2018 compared with the three months ended June 30, 2017
decreased $121,758 or 31.3%. The decrease is related to a decrease in employee salaries and benefits of $58,941 due to a decrease
in employer taxes and employee benefits due the Company reducing costs in 2018 and deferring the payment of salaries and wages
in order to preserve cash and resources to raise additional capital for the Company. The Company also reduced legal and other
professional fees as a result of the Company’s decision to suspend SEC filings beginning in 2017 of approximately $103,000.
The decrease in expense was offset by an increase in consulting costs of approximately $80,000 related to raising capital funds
for the ongoing operations of the Company. Selling,
general and administrative expenses for the six-month period ended June 30, 2018 compared with the three- month period ended June
30, 2017 decreased $409,693 or 44.9%. The decrease is related to a reduction in employee salaries and benefits of $129,687 due
to a decrease in headcount, employer related payroll taxes and health benefit costs and a decrease of $383,144 related to reduced
legal and other professional fees as a result of the Company’s decision to suspend SEC filings and other legal costs related
to the development of non-core products, decrease in sales and marketing expenses, insurance costs and other general office expenses
as the Company reduced all discretionary spending. These decreases were offset by an increase in consulting costs of approximately
$105,435 to raise capital funds for the ongoing operations and development of Generx. Interest
expense increased for the three-month period ended March 31, 2018 compared to the three-month period ended March 31, 2017 by $4,389
or 93.2% and by $10,359 or 155.4% for the six-month period ended June 30, 2018 compared to the six-month period ended June 30,
2017 due to an increase in the note payable during the third and fourth quarter of 2017 of approximately $155,000. For
the Three Months and Nine Months Ended September 30, 2018 compared to the Three Months and Nine Months Ended September 30, 2017 The
following tables sets forth our results of operations for the three-month period and Nine-month ended September 30, 2018 and 2017,
and the relative dollar and percentage change between the two periods.
Change
(2018
to 2017) Change (2018
to 2017) Research
and development expenses for the three-month period ended September 30, 2018 compared to the three- month period ended September
30, 2017 increased by $5,171 or 8.4% which is primarily related to an increase in the cost of Generx product storage and other
clinical supplies of $3,525 and an increase in other miscellaneous expenses of $1,218. Research
and development expenses for the nine-month period ended September 30, 2018 compared to the nine-month period ended September
30, 2017 decreased $65,957 or 25.5% to a decrease in employee benefit costs as the employer portion of health benefits was reduced
by $7,464 and a reduction of clinical trial expenses of $57,302 as the Company stopped research and development activity on non-core
technology product and re-directed resources to the development of Generx and capital funding for the Company’s ongoing
operations. Selling,
general and administrative expenses for the three-month period ended September 30, 2018 compared to the three-month period ended
September 30, 2017 decreased $143,894 or 41% due to a decrease in salary and benefit costs as the two of the Company’s
employees moved from full-time to part-time on a temporary basis and there was a reduction in headcount by one employee. Selling,
general and administrative expenses for the nine-month period ended September 30, 2018 compared to the nine-month period ended
September 30, 2017 decreased $553,587 or 43.8% due to a decrease in salary and benefit costs of $173,400 as the Company’s
overall headcount was reduced by one employee and two employees who moved from full-time employees to part-time
employees, reduction of legal and professional fees of approximately $460,000 as a result of the Company’s decision to suspend
SEC filings, decrease in sales and marketing expenses, insurance costs and other general office expenses as the Company reduced
all discretionary spending. These decreases were offset by an increase in consulting costs of approximately $80,000 to raise capital
funds for the ongoing operations and development of Generx. During
the three-month period and nine-month period ended September 2018, the company recognized a gain on sale of Excellagen technology
to Olaregen in the amount of $650,000, which also represented the cash proceeds on the sale. Interest
expense increased $6,189 or 96.5% for the three-month period ended September 30, 2018 compared to the three-month period ended
September 30, 2017 and $16,548 or 126.5% for the six-month period ended September 30, 2018 compared to the six-month period ended
September 30, 2017 as a result of an increase of $130,000 in the note payable in the fourth quarter of 2017, thereby increasing
the interest expense. For
the Three Months and Six Months Ended June 30, 2017 compared to the Three Months and Six Months Ended June 30, 2016 The
following tables sets forth our results of operations for the three-month period and
Three
Months June
30, Change (2017
to 2016) Six
Months June
30, Change (2017
to 2016) Research
and development expense decreased by $12,681 or 16.1% for the three-month period ended June 30, 2017 compared to the three-month
period ended June 30, 2016 as the Company started to reduce discretionary spending in 2017 in order to conserve cash and focus
Selling,
general and administrative expenses decreased for the three-month period ended June 30, 2017 compared to the three-month period
ended June 30, 2016 by $142,077 or 26.7% as a result of the Company decision to suspend SEC filings, therefore reducing legal
and professional costs and reducing discretionary spending, therefore reducing sales and marketing expenses and insurance costs. Interest
expense increased $1,816 or 62.8% for the three-month period ended June 30, 2017 compared to June 30, 2016 and by $1,200 for the
six-month period ended June 30, 2018 compared to June 30, 2017 as a result of an increase in the note payable in 2017, therefore
increasing the accrued interest each month on the note payable. For
the Three Months and Nine Months Ended September 30, 2017 compared to the Three Months and Nine Months Ended September 30, 2016 The
following tables sets forth our results of operations for the three-month period and Nine-month ended September 30, 2017 and 2016,
and the relative dollar and percentage change between the two periods.
Change (2017
to 2016) Change (2017
to 2016)
Research
and development expense decreased for the three-month period ended September 30, 2017 compared to September 30, 2016 by $67,427
or 52.4% and decreased for the nine-month period ended September 30, 2017 compared to the nine-month period ended September 30,
2016 by $32,680 or 11.2% as a result of a reduction in clinical trial expenses, supplies and other related discretionary expenses
as the Company started to focus resources to raising capital for the Company’s on-going operations and restructure research
and development activities away from non-core products to Generx, the Company’s core product. Selling,
general and administrative expenses decreased $536,068 or 60.4% for the three-month period ended September 30, 2017 compared to
the three-month period ended September 30, 2016 and decreased $499,250 or 28.3% for the nine-month period ended September 30,
2017 compared to the nine-month period ended September 30, 2016, due to a decrease in legal and professional expenses due to the
Company’s decision to suspend SEC filings, while the Company focused efforts on raising capital for the ongoing operations
of the Company and due to a decrease in all discretionary spending in 2017. Interest
expense decreased $999 or 13.5% for the three-month period ended September 30, 2017 compared to the three-month period ended September
30, 2016 as a result of a lower interest rate on the outstanding interest-bearing payables of the Company and increased by $3,202
for the nine-month period ended September 30, 2017 compared to As
a smaller reporting company, we are not required to provide the information required by this item.
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM To
the Shareholders and the Opinion
on the Financial Statements We
have audited the accompanying consolidated balance sheets of Gene Biotherapeutics, Inc. and Subsidiaries (the Explanatory
Paragraph – Going Concern The
accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern.
As more fully described in Note 1, the Company Basis
for Opinion These
consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an
opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered
with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with
respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities
and Exchange Commission and the PCAOB. We
conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit
to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether
due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over
financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting
but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting.
Accordingly, we express no such opinion. Our
audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether
due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis,
evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting
principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial
statements. We believe that /s/
Marcum LLP
Marcum
LLP We
have served as the Company’s auditor since 2012. Houston,
TX April 23, 2021 GENE
BIOTHERAPEUTICS, INC. AND SUBSIDIARIES Series
A Convertible Preferred stock, $0.0001 par value; 40,000,000 shares authorized; issued
and outstanding 790 on December 31, 2019, 800 on December 31, 2018 and 811 on December
31, 2017, with liquidation preferences of $790,000, $800,000, and 811,000. GENE
BIOTHERAPEUTICS, INC. AND SUBSIDIARIES CONSOLIDATED
STATEMENTS OF OPERATIONS
Year
Ended December 31,
2019
2018
2017
Cash
$ 400
$ 82,115
$ 48,989
Other Current Assets
32,395
18,965
25,000
Accounts Payable
967,126
1,857,951
1,870,215
Other Current Liabilities
3,795,863
3,932,835
3,485,440
Working Capital (Deficiency)
(4,730,194 )
(5,689,706 )
(5,281,666 )
●
We
entered into several agreements with employees, former employees, and vendors to restructure claims reducing the amount of
our accounts payable and our other current liabilities and/or extending the payment terms until after commercialization and
Generx products sales commence. -42-
●
In
May 2020 we secured $
Year
Ended December 31,
2019
2018
2017
Net cash generated from
(used in) operating activities
$ (137,162 )
$ (451,503 )
$ (1,062,388 )
Net cash generated from investing activities
—
650,000
—
Net cash generated
from (used in) financing activities
55,447
(165,371 )
180,980
Net increase
(decrease) in cash and cash equivalents
(81,715 )
33,126
(881,408 ) -43-
Three
Months
March 31,
2019
2018
($)
%
Operating Expenses:
Research and development
$ 63,379
$ 62,767
612
1.0 %
Selling, general
and administrative
179,599
234,630
(55,031 )
(23.5 )%
Total Operating
Expenses
242,978
297,397
(54,419 )
(18.3 )%
Loss from Operations
(242,978 )
(297,397 )
54,419
(18.3 )%
Other Income (Expense):
Interest Expense
(10,129 )
(7,927 )
(2,202 )
27.8 %
Total
Other Income (Expense)
(10,129 )
(7,927 )
(2,202 )
27.8 %
Net Loss
(253,107 )
(305,324 )
52,217
(17.1 )%
Net Loss attributable
to the non-controlling interest
(26,738 )
(29,351 )
2,614
(8.9 )%
Net Loss attributable
to the controlling interest
(226,369 )
(275,973 )
49,603
(18.0 )%
Three
Months
June 30,
Six
Months
June 30,
2019
2018
($)
%
2019
2018
($)
%
Operating
Expenses:
Research
and development
$ 60,355
$ 63,049
(2,694 )
(4.3 )%
$ 123,734
$ 125,816
(2,082 )
(1.7 )%
Selling,
general and administrative
180,473
267,629
(87,156 )
(32.6 )%
360,072
502,259
(142,187 )
(28.3 )%
Total
Operating Expenses
240,828
330,678
(89,850 )
(27.2 )%
483,806
628,075
(144,269 )
(23.0 )%
Loss
from Operations
(240,828 )
(330,678 )
89,850
(27.2 )%
(483,806 )
(628,075 )
144,269
(23.0 )%
Other
Income (Expense):
Interest
Expense
(10,504 )
(9,097 )
(1,406 )
15.5 %
(20,633 )
(17,025 )
(3,608 )
21.2 %
Total
Other Income (Expense)
(10,504 )
(9,097 )
(1,406 )
15.5 %
(20,633 )
(17,025 )
(3,608 )
21.2 %
Net
Loss
(251,332 )
(339,775 )
88,442
(26.0 )%
(504,440 )
(645,099 )
140,659
(21.8 )%
Net
Loss attributable to the non-controlling interest
(24,963 )
(27,568 )
2,604
(9.4 )%
(51,701 )
(56,919 )
5,218
(9.2 )%
Net
Loss attributable to the controlling interest
(226,369 )
(312,207 )
85,838
(27.5 )%
(452,739 )
(588,180 )
135,441
(23.0 )% -44-
Three
Months
September 30,
Nine
Months
September 30,
2019
2018
($)
%
2019
2018
($)
%
Operating Expenses:
Research
and development
$ 61,443
$ 66,442
(4,999 )
(7.5 )%
$ 185,177
$ 192,258
(7,081 )
(3.7 )%
Selling,
general and administrative
114,047
207,279
(93,232 )
(45.0 )%
474,119
709,538
(235,419 )
(33.3 )%
Total
Operating Expenses
175,490
273,721
(134,216 )
(35.9 )%
659,296
901,796
(242,500 )
(26.9 )%
Gain
on sale of assets and technology
—
(650,000 )
650,000
(100.0 )%
—
(650,000 )
650,000
(100.0 )%
Income
(Loss)from Operations
(175,490 )
376,279
(515,784 )
(146.6 )%
(659,296 )
(251,796 )
(407,500 )
161.8 %
Other
Income (Expense):
Gain
on account payable forgiveness
35,985
—
—
100.0 %
35,985
—
35,985
100.0 %
Interest
Expense
(10,952 )
(12,603 )
(1,651 )
(13.1 )%
(31,585 )
(29,628 )
(1,957 )
6.6 %
Total
Other Income (Expense)
25,033
(12,603 )
37,636
(298.6 )%
(4,400 )
(29,628 )
34,028
(114.9 )%
Net
Loss
(150,457 )
363,676
(514,132 )
(141.4 )%
(654,896 )
(281,423 )
(373,473 )
132.7 %
Net
Loss attributable to the non-controlling interest
(19,790 )
(31,675 )
11,886
(37.5 )%
(71,490 )
(88,594 )
17,104
(19.3 )%
Net
Loss attributable to the controlling interest
(130,667 )
395,351
(526,018 )
(133.1 )%
(583,406 )
(192,829 )
(390,577 )
202.6 % -45-
Three
Months
March 31,
2018
2017
($)
%
Operating Expenses:
Research and development
$ 62,767
$ 130,762
(67,995 )
(52.0 )%
Selling, general
and administrative
234,630
522,565
(287,935 )
(55.1 )%
Total Operating
Expenses
297,397
653,327
(355,930 )
(54.5 )%
Loss from Operations
(297,397 )
(653,327 )
355,930
(54.5 )%
Other Income (Expense):
Interest Expense
(7,927 )
(1,956 )
(5,970 )
305.1 %
Total
Other Income (Expense)
(7,927 )
(1,956 )
(5,970 )
305.1 %
Net Loss
(305,324 )
(655,283 )
349,959
(53.4 )%
Net Loss attributable
to the non-controlling interest
(29,351 )
(75,160 )
45,809
(60.9 )%
Net Loss attributable
to the controlling interest
(275,973 )
(580,123 )
304,150
(52.4 )% -46-
Three
Months
June 30,
Six
Months
June 30,
2018
2017
($)
%
2018
2017
($)
%
Operating
Expenses:
Research
and development
$ 63,049
$ 66,182
(3,133 )
(4.7 )%
$ 125,816
$ 196,944
(71,128 )
(36.1 )%
Selling,
general and administrative
267,629
389,387
(121,758 )
(31.3 )%
502,259
911,952
(409,693 )
(44.9 )%
Total
Operating Expenses
330,678
455,569
(124,891 )
(27.4 )%
628,075
1,108,896
(480,821 )
(43.4 )%
Loss
from Operations
(330,678 )
(455,569 )
(124,891 )
(27.4 )%
(628,075 )
(1,108,896 )
480,821
(43.4 )%
Other
Income (Expense):
Interest
Expense
(9,097 )
(4,709 )
(4,389 )
93.2 %
(17,025 )
(6,666 )
(10,359 )
155.4 %
Total
Other Income (Expense)
(9,097 )
(4,709 )
(4,389 )
93.2 %
(17,025 )
(6,666 )
(10,359 )
155.4 %
Net
Loss
(339,775 )
(460,278 )
120,503
(26.2 )%
(645,099 )
(1,115,561 )
470,462
(42.2 )%
Net
Loss attributable to the non-controlling interest
(27,568 )
(42,785 )
15,217
(35.6 )%
(56,919 )
(117,945 )
61,026
(51.7 )%
Net
Loss attributable to the controlling interest
(312,207 )
(417,493 )
105,286
(25.2 )%
(588,180 )
(997,616 )
409,436
(41.0 )% -47-
Three
Months
September 30,
Nine
Months
September 30,
2018
2017
($)
%
2018
2017
($)
%
Operating
Expenses:
Research
and development
$ 66,442
$ 61,271
5,171
8.4 %
$ 192,258
$ 258,215
(65,957 )
(25.5 )%
Selling,
general and administrative
207,279
351,173
(143,894 )
(41.0 )%
709,538
1,263,125
(553,587 )
(43.8 )%
Total
Operating Expenses
273,721
412,444
(138,723 )
(33.6 )%
901,796
1,521,340
(619,545 )
(40.7 )%
Gain
on sale of assets and technology
(650,000 )
—
650,000
100.0 %
(650,000 )
—
(650,000 )
100.0 %
Income
(Loss)from Operations
(376,279 )
(412,444 )
36,165
(8.8 )%
(251,796 )
(1,521,340 )
1,269,545
(83.4 )%
Other
Income (Expense):
Interest
Expense
(12,603 )
(6,414 )
(6,189 )
96.5 %
(29,627 )
(13,080 )
(16,548 )
126.5 %
Total
Other Income (Expense)
(12,603 )
(6,414 )
(6,189 )
96.5 %
(29,627 )
(13,080 )
(16,548 )
126.5 %
Net
income(Loss)
363,676
(418,858 )
782,534
(186.8 )%
(281,423 )
(1,534,420 )
1,252,997
(81.7 )%
Net
Loss attributable to the non-controlling interest
(31,675 )
(43,536 )
11,861
(27.2 )%
(88,594 )
(161,482 )
72,888
(45.1 )%
Net
Loss attributable to the controlling interest
395,351
(375,322 )
770,673
(205.3 )%
(192,829 )
(1,372,938 )
1,180,109
(86.0 )% -48-
2017
2016
($)
%
2017
2016
($)
%
Operating
Expenses:
Research
and development
$ 66,182
$ 78,863
(12,681 )
(16.1 )%
$ 196,944
$ 162,197
34,747
21.4 %
Selling,
general and administrative
389,387
531,464
(142,077 )
(26.7 )%
911,952
872,133
39,819
2.0 %
Total
Operating Expenses
455,569
610,327
(154,758 )
(25.4 )%
1,108,896
1,034,330
74,566
7.2 %
Loss
from Operations
(455,569 )
(610,327 )
154,758
(25.4 )%
(1,108,896 )
(1,034,330 )
(74,566 )
7.2 %
Other
Income (Expense):
Interest
Expense
(4,709 )
(2,893 )
1,816
62.8 %
(6,665 )
(5,466 )
1,200
22.0 %
Total
Other Income (Expense)
(4,709 )
(2,893 )
1,816
62.8 %
(6,665 )
(5,466 )
1,200
22.0 %
Net
Loss
(460,278 )
(613,220 )
152,942
(24.9 )%
(1,115,561 )
(1,039,796 )
(75,765 )
7.3 %
Net
Loss attributable to the non-controlling interest
(42,785 )
—
(42,785 )
(100.0 )%
(117,945 )
—
(117,945 )
(100.0 )%
Net
Loss attributable to the controlling interest
(417,493 )
—
(417,493 )
(100.0 )%
(997,616 )
—
(997,616 )
(100.0 )%
Three
Months
September 30,
Nine
Months
September 30,
2017
2016
($)
%
2017
2016
($)
%
Operating
Expenses:
Research
and development
$ 61,271
$ 128,698
(67,427 )
(52.4 )%
$ 258,215
$ 290,895
(32,680 )
(11.2 )%
Selling,
general and administrative
351,173
887,241
(536,068 )
(60.4 )%
1,263,125
1,762,375
(499,250 )
(28.3 )%
Total
Operating Expenses
412,444
1,015,939
(603,495 )
(59.4 )%
1,521,340
2,053,270
(531,929 )
(25.9 )%
Loss
from Operations
(412,444 )
(1,015,939 )
603,495
(59.4 )%
(1,521,340 )
(2,053,270 )
531,929
(25.9 )%
Other
Income (Expense):
Interest
Expense
(6,414 )
(7,413 )
999
(13.5 )%
(13,080 )
(9,878 )
3,202
32.4 %
Total
Other Income (Expense)
(6,414 )
(7,413 )
999
(13.5 )%
(13,080 )
(9,878 )
3,202
32.4 %
Net
Loss
(418,858 )
(1,023,352 )
604,494
(59.1 )%
(1,534,420 )
(2,063,148 )
528,728
(25.6 )%
Net
Loss attributable to the non-controlling interest
(43,536 )
(19,460 )
(24,076 )
100.0 %
(161,482 )
(19,460 )
(161,482 )
100.0 %
Net
Loss attributable to the controlling interest
(375,322 )
(1,003,892 )
(375,322 )
100.0 %
(1,372,938 )
(2,043,688 )
(1,372,938 )
100.0 %
Deemed
dividend on preferred stock
—
782,879
(782,879 )
(100.0 )%
—
782,879
(782,879 )
(100.0 )%
Net
loss applicable to common stockholders
—
(1,806,231 )
1,806,231
(100.0 )%
—
(2,846,027 )
2,846,027
(100.0 )% -61-
December
31,
2019
2018
2017
Assets
Current
assets:
Cash
$ 400
$ 82,115
$ 48,989
Prepaid
expenses and other assets
32,395
18,965
25,000
Total
current assets
32,795
101,080
73,989
Property
and equipment, net
2,640
60,815
124,017
Other
long—term assets
—
—
11,767
Total
other assets
2,640
60,815
135,784
Total
assets
$ 35,435
$ 161,895
$ 209,773
Liabilities
and Stockholders’ Deficit
Current
liabilities:
Accounts
payable
$ 967,126
$ 1,857,951
$ 1,870,215
Accrued
liabilities
2,796,689
2,856,634
2,244,517
Advances
from officer
725,425
825,187
1,019,804
Notes
payable-Current
273,749
240,749
211,503
Deferred
rent
—
10,265
9,616
Total
current liabilities
4,762,989
5,790,786
5,355,655
Notes
payable-Long term
122,209
—
—
Deferred
rent
—
—
10,265
Total
liabilities
4,885,198
5,790,786
5,365,920
Commitments
and contingencies
—
—
—
Stockholders’
deficit:
—
—
—
Common
stock, $0.0001 par value; 200,000,000 shares authorized; issued and outstanding 14,489,399 on December 31, 2019, 14,433,843
on December 31, 2018 and 14,373,544 on December 31, 2017
1,449
1,444
1,438
Common
stock issuable
600,000
600,000
600,000
Additional
paid-in capital
114,020,581
114,020,586
113,940,592
Accumulated
deficit
(118,912,290 )
(119,778,965 )
(119,344,084 )
Total
controlling interest
(4,290,260 )
(5,156,935 )
(4,802,054 )
Non-controlling
interest
(559,503 )
(471,956 )
(354,093 )
Total
stockholders’ deficit
(4,849,763 )
(5,628,891 )
(5,156,147 )
Total
liabilities and stockholders’ deficit
$ 35,435
$ 161,895
$ 209,773 -62-
| Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Operating expenses | ||||||||||||
| Research and development | $ | 243,453 | $ | 255,394 | $ | 344,976 | ||||||
| Selling, general and administrative | 593,549 | 907,836 | 1,781,309 | |||||||||
| Total operating expenses | 837,002 | 1,163,230 | 2,126,285 | |||||||||
| Gain on sale of assets and technology | — | (650,000 | ) | (50,000 | ) | |||||||
| Income (loss) from operations | (837,002 | ) | (513,230 | ) | (2,076,285 | ) | ||||||
| Other Income (expenses): | ||||||||||||
| Gain on forgiveness of accounts payable | 1,659,917 | — | — | |||||||||
| Interest expense | (43,787 | ) | (39,514 | ) | (20,219 | ) | ||||||
| Total other Income (expenses) | 1,616,130 | (39,514 | ) | (20,219 | ) | |||||||
| Net Income (loss) | 779,128 | (552,744 | ) | (2,096,504 | ) | |||||||
| Net Income (loss) attributable to the non-controlling interest | (87,547 | ) | (117,863 | ) | (202,362 | ) | ||||||
| Net (Loss) attributable to the controlling interest | $ | 866,675 | $ | (434,881 | ) | $ | (1,894,142 | ) | ||||
| Net loss per share—basic and diluted | $ | 0.05 | $ | (0.03 | ) | $ | (0.13 | ) | ||||
| Weighted average number of common shares outstanding – basic and diluted | 14,485,604 | 14,415,418 | 14,290,416 | |||||||||
| -63- |
GENE BIOTHERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS
YEARS
ENDED DECEMBER
| Controlling Interest | ||||||||||||||||||||||||||||||||||||
| Series A | ||||||||||||||||||||||||||||||||||||
| Convertible | Common | Additional | Controlling | Non- | ||||||||||||||||||||||||||||||||
| Common Stock | Preferred Stock | Stock | Paid-In- | Interest | Controlling | Deficit | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Issuable | Capital | Deficit | Interest | Total | ||||||||||||||||||||||||||||
| Balance—December 31, 2016 | 13,723,544 | $ | 1,373 | 928 | $ | — | $ | 600,000 | $ | 113,710,631 | $ | (117,449,942 | ) | $ | (151,731 | ) | $ | (3,289,669 | ) | |||||||||||||||||
| Issuance of common stock on conversion of preferred stock | 650,000 | 65 | (117 | ) | — | (65 | ) | — | ||||||||||||||||||||||||||||
| Stock-based compensation | 803 | 803 | ||||||||||||||||||||||||||||||||||
| Issuance of warrants to purchase common shares | 229,223 | 229,223 | ||||||||||||||||||||||||||||||||||
| Non-controlling interest | (202,362 | ) | (202,362 | ) | ||||||||||||||||||||||||||||||||
| Net loss | (1,894,142 | ) | (1,894,141 | ) | ||||||||||||||||||||||||||||||||
| Balance—December 31, 2017 | 14,373,544 | $ | 1,438 | 811 | $ | — | $ | 600,000 | $ | 113,940,592 | $ | (119,344,084 | ) | $ | (354,093 | ) | $ | (5,156,146 | ) | |||||||||||||||||
| Issuance of common stock on conversion of preferred stock | 60,299 | $ | 6 | (11 | ) | $ | $ | (6 | ) | — | ||||||||||||||||||||||||||
| Issuance of warrants to purchase common shares | 80,000 | 80,000 | ||||||||||||||||||||||||||||||||||
| Non-controlling interest | (117,863 | ) | (117,863 | ) | ||||||||||||||||||||||||||||||||
| Net loss | (434,881 | ) | (434,881 | ) | ||||||||||||||||||||||||||||||||
| Balance—December 31, 2018 | 14,433,843 | $ | 1,444 | 800 | — | $ | 600,000 | $ | 114,020,586 | $ | (119,778,965 | ) | $ | (471,956 | ) | $ | (5,628,890 | ) | ||||||||||||||||||
| Issuance of common stock on conversion of preferred stock | 55,556 | 5 | (10 | ) | — | (5 | ) | — | — | |||||||||||||||||||||||||||
| Non-controlling interest | — | (87,547 | ) | (87,547 | ) | |||||||||||||||||||||||||||||||
| Net income | 866,675 | 866,674 | ||||||||||||||||||||||||||||||||||
| Balance—December 31, 2019 | 14,489,399 | $ | 1,449 | 790 | $ | — | $ | 600,000 | $ | 114,020,581 | $ | (118,912,290 | ) | $ | (559,503 | ) | $ | (4,849,763 | ) | |||||||||||||||||
| -64- |
GENE BIOTHERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Cash Flows from Operating Activities | ||||||||||||
| Net Income (loss) | $ | 779,128 | $ | (552,744 | ) | $ | (2,096,504 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation | 58,175 | 63,202 | 66,087 | |||||||||
| Deferred rent amortization | (10,265 | ) | (9,616 | ) | (18,883 | ) | ||||||
| Gain on forgiveness of accounts payable | (1,659,917 | ) | — | — | ||||||||
| Stock-based compensation | — | — | 803 | |||||||||
| Warrants issued in exchange for services | — | 80,000 | 229,223 | |||||||||
| Gain on sale of assets and technology | — | (650,000 | ) | (50,000 | ) | |||||||
| Changes in operating assets and liabilities | ||||||||||||
| Prepaid expenses and other assets | (25,971 | ) | 18,576 | 35,427 | ||||||||
| Deposits | 12,541 | (774 | ) | — | ||||||||
| Accounts payable | 77,511 | (12,263 | ) | 197,151 | ||||||||
| Accrued liabilities | (631,636 | ) | 612,116 | 574,308 | ||||||||
| Net cash from (used in) operating activities | (137,162 | ) | (451,503 | ) | (1,062,388 | ) | ||||||
| Cash Flows from Investing Activities | ||||||||||||
| Proceeds on sale or transfer of assets and technology | — | 650,000 | — | |||||||||
| Net cash provided from investing activities | 650,000 | |||||||||||
| Cash Flows from Financing Activities | ||||||||||||
| Proceeds (Payments) from officer’s loan | (99,762 | ) | (194,617 | ) | (30,523 | ) | ||||||
| Proceeds on issuance of notes payable | 155,209 | 29,246 | 211,503 | |||||||||
| Net cash provided by financing activities | 55,447 | (165,371 | ) | 180,980 | ||||||||
| Net increase (decrease) in cash and cash equivalent | (81,715 | ) | 33,126 | (881,408 | ) | |||||||
| Cash and cash equivalents at beginning of year | 82,115 | 48,989 | 930,397 | |||||||||
| Cash and cash equivalents at end of year | $ | 400 | $ | 82,115 | $ | 48,989 | ||||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||||||
| Cash paid for interest | $ | 8,716 | $ | 10,269 | $ | 8,578 | ||||||
| Cash paid for income taxes | $ | — | $ | — | $ | — | ||||||
| Supplemental noncash investing activities: | ||||||||||||
| Transfer of assets and technology in exchange for consulting services | — | — | $ | 50,000 | ||||||||
GENE
BIOTHERAPEUTICS, INC., AND SUBSIDIARIES NOTES
TO CONSOLIDATED FINANCIAL STATEMENTS Note
1. Organization and Liquidity Gene
Biotherapeutics, Inc. was initially incorporated in Delaware in December 2003. The
Company’s current business is focused exclusively on the development of Generx, a gene therapy product candidate targeted
for Liquidity
and Going Concern
As
of December 31, 2019, the Company had $400 in cash and cash equivalents. The Company’s working capital deficit on December
31, 2019 was $4,730,194 and the Company has incurred recurring losses and has an accumulated deficit of $118,912,290.
During the years ended December 31, 2019, 2018 and 2017, the Company used approximately $137,162, 451,303 and used $1,062,388
of cash in its operating activities, respectively. The
Company’s primary source of capital is from proceeds from sales of its equity securities and in 2018 included the sale of
Excellagen generating cash of $650,000 plus potential royalty payment of 10% of all worldwide sales of Excellagen. In
July 2018, we sold our FDA-cleared Excellagen® product to Olaregen for aggregate consideration of up to $4,000,000. At closing,
we received a cash payment of $650,000, the remaining to be paid as royalty payments of 10% of all worldwide sales of Excellagen
outside of China, up to an additional $3,350,000. We retained rights to manufacture, market and sell Excellagen in Greater China,
The Russian Federation, and the CIS. In
April 2020, after the period covered by this report, we transferred our residual rights in Excellagen to Shanxi in exchange for
the release of any rights or claims in ownership interest in Gene Biotherapeutics. As a result, we no longer have an interest
in Excellagen, other than the right to receive royalty payments from Olaregen totaling up to $3,350,000, based on monthly net
sales of Excellagen worldwide, excluding Greater China, the Russian Federation, and countries in the CIS. In connection with this
transaction, Shanxi agreed to apply previously funded $600,000 subscription payment in exchange for the rights to Excellagen in
the Greater China, the Russian Federation and countries in the CIS, and Shanxi released any future rights or claims against us. On
April 10, 2020, after the period covered by this report, our Angionetics, Inc. subsidiary entered into the Shanxi License Agreement,
granting Shanxi certain license rights with respect to our Generx product candidate. The distribution and license rights commence
only after we obtain U.S. FDA approval for marketing and sale of Generx in the United States. The license rights include (a) a
non-exclusive right to manufacture Generx products in China, and (b) an exclusive right to market and sell Generx products in
Singapore, Macau, Hong Kong, Taiwan, any other municipality other than mainland China where Chinese (Mandarin or Cantonese) is
the common language, the Russian Federation, and the CIS (i.e., Armenia, Azerbaijan, Belarus, Kazakhstan, Kyrgyzstan, Moldova,
Tajikistan, Turkmenistan, and Uzbekistan). The Shanxi License Agreement provides for a royalty ranging from 5% up to 10% based
on the level of annual net sales of the In
advance of -65- -66-
The
Company anticipates that negative cash flows from operations will continue for the foreseeable future and
The Company’s principal business objectives are to advance the independent monetization and funding activities of our core products and technologies, with our Angionetics Inc. subsidiary being focused on the Generx angiogenic gene therapy product candidate.
The
accompanying consolidated financial statements have been prepared in conformity with U.S. GAAP, which contemplates our continuation
as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business
Note 2—Summary of Significant Accounting Policies
Basis of Presentation
The
accompanying consolidated financial statements have been prepared in accordance with U.S. GAAP and the rules and regulations of
the
Fair Value of Financial Instruments
The
carrying amounts of cash and cash equivalents,
Use of Estimates and assumptions and critical accounting estimates and assumptions
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
The
most significant estimates and critical accounting policies involve valuing warrants using option pricing models and determination
of the
Actual
results could differ from these estimates. Management’s estimates and assumptions are reviewed regularly, and the effects
of revisions are reflected in the consolidated financial statements
The
consolidated financial statements include the accounts of
| -67- |
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of three months or less when purchased to be cash equivalents. The Company had no cash equivalents for each of the years ended December 31, 2019, 2018 and 2017.
Concentration of Credit Risk
Financial
instruments that potentially subject us to significant concentrations of credit risk consist of cash and cash equivalents. At
times, our cash and cash equivalents may be uninsured or in deposit accounts that exceed the Federal Deposit Insurance Corporation
Property and Equipment, net
Property
and equipment are stated at cost and include equipment, installation costs and materials less accumulated depreciation and amortization.
Depreciation is calculated on a straight-line basis over the estimated useful lives of the assets. Estimated useful lives of the
assets range from 3 to 5 years. Leasehold improvements are amortized over the lesser of the useful lives or the term of the respective
lease. Expenditures for maintenance and repairs, which do not extend the useful life of the assets, are charged to expense as
incurred. Gains or losses on disposal of property and equipment are reflected in general and administrative expenses in the statement
of operations.
Impairment of Long-Lived Assets
The
Company assesses its property and equipment
Preferred Stock
Revenue Recognition
The Company’s revenue recognition accounting policy until December 31, 2017, prior to the adoption of the new revenue standard, FASB issued Accounting Standards (ASU 606- “Revenue from Contracts with Customers”).
The Company recognizes revenue from product sales when (i) persuasive evidence of an arrangement exists, (ii) delivery has occurred, (iii) the sales price is fixed or determinable, and (iv) collectability is reasonably assured. These criteria are typically met when the risk of ownership and title passes to our customers. Up to the period ended December 31, 2017, the Company has not recognized revenue from product sales.
The Company’s revenue recognition accounting policy from January 1, 2018, following the adoption of the new revenue standard, ASU 606.
The Company’s products have not reached commercialization, accordingly revenue from product sales have not been recognized. For arrangements that include sales-based royalties, the Company recognizes revenue based on an assessment of the probability of achievement. There is considerable judgement involved in determining whether it is probable that royalties will be collected. At the end of each subsequent reporting period, the Company reevaluates the probability of achievement and if necessary, adjusts the estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenues and earnings in the period of adjustment. To date, the Company has not recognized revenue from product sales or for royalties.
| -68- |
Research and Development
Research
and development
Income Taxes
Income
taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future
tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities
and their respective tax bases
The
Company has adopted the provisions of ASC 740-10, which clarifies the accounting for uncertain tax positions. ASC 740-10 requires
that the Company recognize the impact of a tax position in its financial statements if the position is more likely than not to
be sustained upon examination
The
Company’s policy is to recognize interest and penalties
When
tax returns are filed, there may be uncertainty about the merits of positions taken or the amount of the position that would be
ultimately sustained.
Tax
positions that meet the more likely than not recognition threshold is measured at the largest amount of tax benefit that is more
than 50 percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefit associated
with tax positions taken that exceed the amount measured as described above should be reflected as a liability for uncertain tax
benefits in the accompanying balance sheet along with any associated interest and penalties that would be payable to the taxing
authorities upon examination. The Company believes our tax positions are all more likely than not to be upheld upon examination.
As such, the Company has not
Common Stock Purchase Warrants
The
Company accounts for the issuance of common stock purchase warrants issued in connection with capital financing transactions
Earnings (Loss) Per Common Share
Basic
earnings (loss) per common share is computed by dividing
| -69- |
Stock-Based Compensation
The
Company recognizes the fair value of all share-based payment awards in the
The
Company estimated the fair value of an option or warrant award on the date of grant using the Black
Determining
the appropriate fair value model and calculating the fair value of equity
The
Company recognized total stock-based compensation expense
Recent Accounting Pronouncements
In
May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers (Topic 606). The new standard is based on
the principle that revenue should be recognized in an amount that reflects the consideration to which the entity expects to be
entitled in exchange for the transfer of promised goods or services. ASU 2014-09 and all subsequent amendments (collectively,
the “new standards”) may be applied using either the full retrospective method, in which case the standard would be
applied to each prior reporting period presented, or the modified retrospective method, in which case the cumulative effect of
applying the standard would be recognized at the date of initial application. The Company has adopted the standards beginning
this first quarter of 2018 using the modified retrospective method. Overall, the timing or amounts related to the revenue recognition
under the new standards did not differ from our previously applied revenue recognition policy. Our product revenues are recognized
at a point in time, which is when control transfers to the customer. The Company has made an accounting policy election to treat
shipping and handling activities that occur after the customer obtains control of the goods as fulfillment costs. There was no
cumulative effect of applying the new standards as of the adoption date on January 1, 2018. For
arrangements that include sales-based royalties, including milestone payments based on the level of sales, the Company recognizes
revenue based on an assessment of the probability of achievement of the milestones. There is considerable judgement involved in
determining whether it is probable that a significant revenue reversal would not occur. At the end of each subsequent reporting
period, the Company revaluates the probability of achievement of all milestones subject to constraint and, if necessary, adjusts
the estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect
revenues and earnings in the period of adjustment. In
January 2016, the FASB issued ASU 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities, which
makes targeted improvements to the accounting for, and presentation and disclosure of financial statements. ASU No. 2016-01 requires
that most equity investments be measured at fair value, with changes in fair value recognized in net income. The pronouncement
also eliminated certain fair value disclosure requirements previously required under GAAP. The Company adopted this standard on
January 1, 2019, which did not have a material impact on the financial statements. In
February 2016, the FASB issued Accounting Standards Update (“ASU”) 2016-02, Leases. Under this new guidance,
at the commencement date, lessees will be required to recognize (i) a lease liability, which is a lessee’s obligation to
make lease payments arising from a lease, measured on a discounted basis and (ii) a right-of-use asset, which is an asset that
represents the lessee’s right to use, or control the use of, a specified asset for the lease term. This guidance is not
applicable for leases with a term of 12 months or less. The Company will adopt the new guidance in the first quarter of fiscal
2020 and expect to use the modified retrospective approach and to elect certain practical expedients, with the cumulative effect
of applying the new guidance recognized as an adjustment to opening retained earnings in the year of adoption. The adoption is
not expected to have a material impact on the Company’s consolidated financial statements. In
August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and
Cash Payments. This standard makes eight targeted changes to how cash receipts and cash payments are presented and classified
in the statement of cash flows. The Company adopted this standard beginning January 1, 2019. The impact of the adoption of ASU
No. 2016-15 did not have a material impact on the financial statements. In
June 2018, the FASB issued ASU No. 2018-07, Compensation - Stock Compensation (Topic 718): Improvements to -70-
In
August 2018, the FASB issued
Note 3—Disposal of Long-Lived Assets
In October 2017, the Company entered into an Agreement, for a minimum six-month period, with Landmark, a third-party corporate finance advisory and strategy consulting firm specializing in the life sciences and healthcare industries. Under the terms of the agreement, Landmark will assist with the sale of the Company’s non-strategic assets, including the Excellagen assets, the Company’s preferred stock equity interest in Healthy Brands Collective and LifeAgain advanced medical data analytics asset group. Landmark is also engaged to assist the Company with identifying potential parties and evaluating proposals for raising development equity capital and with opportunities to market and monetize the Generx technology platform. In exchange for the services rendered, the Company paid Landmark non-monetary consideration which included the assignment of the Company’s interest in Healthy Brands Collective, Nutra-Apps, and LifeAgain to Landmark covering Landmark’s unilateral right to effect transactions focused on the economic monetization or business mobilization by Landmark as initial retainer consideration. The Company shall have a right to receive 25% of any deal consideration that is received by Landmark for any amounts paid in excess of $50,000 during the 12 months following the execution of the Agreement. The Company determined the fair value of the consideration to be $50,000, representing the retainer fee that has been settled by transferring these group of assets. Accordingly, in October 2017, the Company recognized a gain on the transfer of assets in the amount of $50,000 and a prepaid asset, which was amortized to consulting expenses for the period from October 2017 to March 31, 2018.
In July 2018, Activation Therapeutics, the Company’s wholly owned subsidiary, entered into an asset purchase agreement (the “Excellagen Agreement”) and sold our Excellagen® flowable dermal matrix for wound healing technology to Olaregen for aggregate consideration of up to $4,000,000. At closing, the Company received a cash payment of $650,000, and is entitled to receive quarterly royalty payments equal to 10% of all worldwide sales of Excellagen, up to an additional $3,350,000. In addition, under the Excellagen Agreement, (1) the Company is entitled to receive a 2% royalty on net revenue of Excellagen-based products utilizing exosomes, (2) the Company is entitled to 25% of any consideration that may include, but not be limited to, the licensing fees, sale, strategic partnering, and royalties, that may be received from Olaregen.
| -71- |
The Company recognized a gain on the sale of the Excellagen® assets in the amount of $650,000 during its quarter ended September 30, 2018. The remaining $3,350,000 in additional consideration is payable at a rate of 10% on quarterly worldwide net sales recognized by Olaregen of Excellagen® product and will be recognized as a gain on sale of Excellagen assets in the period that Olaregen reports sales and collection is reasonably assured.
On April 10, 2020, subsequent to the date of this report, the Company’s Activation Therapeutics, Inc. subsidiary entered into the Shanxi Assignment Agreement pursuant to which the Company transferred all of its license rights to manufacture, use, market and sell Excellagen to Shanxi. The Company also assigned to Shanxi a Chinese patent that the Company received on Excellagen. As a result, the Company no longer has an interest in Excellagen, other than the right to the royalty payments from Olaregen.
Property and equipment consisted of the following:
| December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Computer and telecommunication equipment | $ | 12,902 | $ | 12,902 | $ | 12,902 | ||||||
| Office equipment | 5,871 | 5,871 | 5,871 | |||||||||
| Office furniture and equipment | 7,396 | 7,396 | 7,396 | |||||||||
| Leasehold improvements | 177,436 | 177,436 | 177,436 | |||||||||
| 203,605 | 203,605 | 203,605 | ||||||||||
| Accumulated depreciation | (200,965 | ) | (142,790 | ) | (79,588 | ) | ||||||
| Property and equipment, net | $ | 2,640 | $ | 60,815 | $ | 124,017 | ||||||
Depreciation
Note 5—Accrued Liabilities
| December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Payroll and benefits | $ | 2,667,717 | $ | 2,114,732 | $ | 1,555,815 | ||||||
| Other | 128,972 | 741,902 | 688,702 | |||||||||
| Total | $ | 2,796,689 | $ | 2,856,634 | $ | 2,244,517 | ||||||
As of December 31, 2019, 2018 and 2017, the Company had unpaid salaries and benefits for current and former employees totaling $2,866,717, $2,288,732, and $1,555,815 which has been offset by cash advances paid to employees totaling $199,000, $174,000 and $nil, respectively.
In January 2020, all affected current and former employees agreed to defer their compensation, less applicable tax withholdings, upon the earliest to occur of (a) the FDA’s approval of Generx for marketing and sale in the U.S.; (b) the EMA approval of Generx for marketing and sale in the European Union and the United Kingdom; (c) the sale of Generx to an independent third party for an aggregate value equal to or greater than $35,000,000; (d) the Company’s entry into a strategic partnership that would facilitate a capital contribution equal to or greater than $35,000,000 for the purpose of supporting the clinical and commercial development of Generx; (e) the Company’s successful completion of a public or private equity offering for the issuance of its common stock equal to $35,000,000; or (f) at such other time, as our board of directors determines that the Company has the financial ability to make such payments without jeopardizing its ability to operate as a going concern.
Note 6—Advances from Related Party-Officer
As of December 31, 2019, 2018, and 2017, $725,425, $825,187, and $1,019,804, respectively, in net Company expenses incurred in the ordinary course of business have been paid by or with cash advanced by Christopher J. Reinhard, the Company’s Chief Executive Officer. These advances are non-interest bearing with no fixed terms of repayment. During the year ended December 31, 2019, 2018, and 2017, the Company repaid $99,762, $194,617, and $30,523, respectively. Subsequent to December 31, 2019, effective June 2020, the Company is repaying the advances from related party-officer in equal monthly installments of $20,000.
| -72- |
Note 7—Notes Payables
As of December 31, 2019, 2018, and 2017 $273,749, $240,749, and $211,503, respectively the company had notes payables and accrued interest outstanding. These notes accrue interest at 15% beginning May 13, 2018 and accrued interest at 10% from inception to May 13, 2018. These notes were accrued with no fix terms of repayment.
On September 10, 2019, the company issued a note payable to Nostrum in exchange for $120,000 in cash. This note bears interest at 6% per annum and the accrued interest and principal is due and payable on September 10, 2021. As of December 31, 2019, the total payable is $122,209 representing principal plus accrued interest.
Note 8—Commitments and Contingencies
Lease Commitments
On
June 23, 2016,
On July 8, 2020, the Company entered a twenty-nine-month lease for approximately 3,039 square feet of office space in San Diego, California commencing on August 1, 2020. The monthly base rent is $6,685 and increases by three percent (3%) on each anniversary of the Commencement Date.
Future annual minimum rental payments under the leases are as follows:
| Year Ending December 31, | Facilities
(Operating Lease) | |||
| 2020 | $ | 74,730 | ||
| 2021 | 81,228 | |||
| 2022 | 83,642 | |||
| Total | $ | 239,600 | ||
Rent
expense including common area expenses, was $
Contingencies
During 2019 and subsequent to the period ended December 31, 2019, the Company has entered into various restructuring efforts including the restructuring of certain payables with its vendors to pay certain amounts due contingent up on the receipt of FDA approval on Generx or contingent up on the FDA approval and commercial sales of Generx. Since it is not determinable when and if Generx will receive FDA approval and the Company will achieve commercial sales, the Company has reflected these re-negotiated amounts due as contingent liabilities where it is not determinable when and if the amounts will ultimately be paid. The total liabilities payable by the Company in the event of FDA approval is $172,449 and an additional amount totaling $225,000 is payable when commercial sales cumulatively reach $100 million for Generx. Since the Company does not know if FDA approval will be received for the Generx product, it is not determinable if and when this payment will be made by the Company. Accordingly, these amounts have been reported as a contingent liability and have not been included in accounts payable and accrued liabilities.
| -73- |
License Fees
Technology License Agreements
In
October 2005, the Company completed a transaction with Schering
As
of October 31, 2019, the outstanding and unpaid amount due and payable under the UC License Agreement totaled $1,006,709. As part
of
As
of November 2019, the Company and the New York University reached an agreement to settle total amounts due under this agreement
for $400,000 payable as follows: (1) $75,000
The Company has not reflected the contingent amounts payable of $397,449 in the Consolidated Balance Sheet as the payable is contingent on FDA approval and commercialization of the product. Since it is not determinable when and if FDA approval will be received, it is not determinable if and when this payment will be made by the Company. Accordingly, these amounts have been reported as a contingent liability. As a result of these settlements, the agreements are deemed terminated and no further amounts and royalties are payable by the Company.
Legal Proceedings
During
the course of our business, the Company is routinely involved in proceedings such as disputes involving goods or services
provided by various third parties, which
Note 9—Income Taxes
The
Company files U. S. federal and state of California income tax returns.
The
Company has not filed its federal and state income tax returns for the years ended December 31, 2019, 2018 and 2017. Net operating
losses for these years will not be available to reduce future taxable income until the returns are filed. The Company expects
to file delinquent tax returns in the upcoming reporting periods and updated values will be disclosed in these following reporting
periods. Assuming these returns are filed, the Company has net operating loss carryforwards for federal income tax purposes of
approximately $109.6 million, $110.7 million, and $ , if unused. The federal net operating loss carryover includes
$258,000 of net operating losses generated in 2018 and later. Federal net operating losses generated from 2018 onwards carryover
indefinitely and may generally be used to offset up to 80% of future taxable income. The Company also has R&D tax credits
available for federal and state purposes of $1.9 million and $2.0 million, respectively. The federal R&D credits will
begin to expire December 31, 2035.
| -74- |
The
ultimate realization of deferred tax assets depends on the generation of future taxable income during the periods in which those
net operating losses are available.
A
valuation
In December 2017, the Tax Cuts and Jobs Act (the “2017 Act”) was enacted. The 2017 Act includes a number of changes to existing U.S. tax laws that impact the Company, most notably a reduction of the U.S. corporate income tax rate from 34 percent to 21 percent for tax years beginning after December 31, 2017. In 2017, the Company recorded provisional amounts for certain enactment-date effects of the act by applying the guidance in Staff Accounting Bulletin No. 118 (“SAB 118”). In 2018 and 2017, the Company recorded $0 net tax expense related to the enactment-date effects of the 2017 Act related to the remeasurement of deferred tax assets and liabilities.
The Company applied the guidance in SAB 118 when accounting for the enactment-date effects of the 2017 Act in 2017 and throughout 2018. On December 31, 2017, the Company had not completed its accounting for all of the enactment-date income tax effects of the 2017 Act under ASC 740, Income Taxes, related to the remeasurement of deferred tax assets and liabilities. On December 31, 2018, the Company has now completed our accounting for all of the enactment-date income tax effects of the 2017 Act and no adjustments were made to the provisional amounts recorded on December 31, 2017.
As of December 31, 2017, the Company remeasured certain deferred tax assets and liabilities based on the rates at which they were expected to reverse in the future (which was generally 21%), by recording a provisional amount of $14.5 million, which was fully offset by valuation allowance. Upon further analysis of certain aspects of the 2017 Act and refinement of our calculations during the 12 months ended December 31, 2018, the Company determined that no adjustment was necessary to our provisional amount.
The
Company’s net deferred tax asset consisted of the following on December
| December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Deferred tax asset: | ||||||||||||
| Net operating loss carryforwards | $ | 28,067,400 | $ | 28,356,900 | $ | 28,284,600 | ||||||
| Deferred compensation | 1,165,300 | 1,165,300 | 1,142,900 | |||||||||
| Depreciation and amortization | 269,300 | 359,600 | 448,900 | |||||||||
| Research and development credits | 3,451,000 | 3,428,700 | 3,408,700 | |||||||||
| Accrued expenses | 830,200 | 668,400 | 435,400 | |||||||||
| Impairment loss | 513,400 | 513,400 | 513,400 | |||||||||
| Other | 216,800 | 216,800 | 300,800 | |||||||||
| Total deferred tax assets | 34,513,400 | 34,709,100 | 34,534,700 | |||||||||
| Less: Valuation allowance | (34,513,400 | ) | (34,709,100 | ) | (34,534,700 | ) | ||||||
| Net deferred tax asset | $ | — | $ | — | $ | — | ||||||
The
income tax provision (benefit) from income taxes consists of the following
| Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Federal | ||||||||||||
| Current | $ | — | $ | — | $ | — | ||||||
| Deferred | 151,422 | (126,818 | ) | 15,182,777 | ||||||||
| State | ||||||||||||
| Current | — | — | — | |||||||||
| Deferred | 44,271 | (47,599 | ) | 643,713 | ||||||||
| Total | 195,693 | (174,417 | ) | 15,826,490 | ||||||||
| Change in valuation allowance | (195,693 | ) | 174,417 | (15,826,490 | ) | |||||||
| Income tax provision (benefit) | $ | — | $ | — | $ | — | ||||||
| -75- |
As
a result of our significant operating loss carry forwards and the corresponding valuation allowance, no income tax benefit was
recorded
| December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Federal income tax rate | 21.0 | % | 21.0 | % | 34.0 | % | ||||||
| State income tax rate, net of federal benefit | 6.3 | % | 8.9 | % | 5.8 | % | ||||||
| Income tax return to tax provision adjustment | — | % | — | % | (45.5 | )% | ||||||
| Impact of U.S. 2017 tax act | — | % | — | % | (763.5 | )% | ||||||
| Other permanent differences | (2.1 | )% | 5.7 | % | 0 .0 | % | ||||||
| Net operating loss expiration | — | % | — | % | (67.8 | )% | ||||||
| Tax credits | (2.6 | )% | 4.6 | % | 1.4 | % | ||||||
| 22.6 | % | 40.1 | % | (835.6 | )% | |||||||
| Change in valuation allowance | (22.6 | )% | (40.1 | )% | 835.6 | % | ||||||
| — | % | — | % | — | % | |||||||
Note 10—Stockholders’ Equity
Common Stock
On
April 4, 2015, the Company entered into a term sheet with Shenzhen Qianhai Taxus Capital Management Co., Ltd. (
Shenzhen Qianhai Taxus held approximately 25% of the Company shares as of December 31, 2019 and the chairman of Shenzhen Qianhai Taxus was also on the Board of Directors and Chairman of the Board of the Company. After the change of control described in the subsequent event note, the chairman resigned from the Board and the ownership of Shenzhen was diluted to less than 10%.
Preferred Stock
Exchange and Redemption Agreement with Sabby Healthcare Volatility Master Fund, Ltd.
On
April 4, 2013,
| -76- |
On
July 22, 2015, the Company entered into an Exchange and Redemption Agreement with Sabby relating to the 1,176 outstanding shares
of Preferred Stock that remained outstanding at that time.
Stock Options and Other Equity Compensation Plans
The
Company had an equity incentive plan that was established in 2005 under which 283,058 shares of
At
December 31,
The
following is a summary of stock option and warrant activities under our equity incentive plan and warrants issued outside of the
plan to employees and
Number of Options or Warrants |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (in years) |
||||||||||
| Balance outstanding, January 1, 2017 | 12,116,334 | $ | 0.73 | 7.67 | ||||||||
| Granted | 1,700,000 | 0.25 | 9.81 | |||||||||
| Exercised | — | — | — | |||||||||
| Cancelled (unvested) | — | — | — | |||||||||
| Expired (vested) | (5,001 | ) | 0.15 | — | ||||||||
| Balance outstanding, December 31, 2017 | 13,811,333 | 0.65 | 7.06 | |||||||||
| Granted | 1,000,000 | 0.25 | 9.31 | |||||||||
| Exercised | — | — | — | |||||||||
| Cancelled (unvested) | — | — | — | |||||||||
| Expired (vested) | — | — | — | |||||||||
| Balance outstanding, December 31, 2018 | 14,811,333 | $ | 0.62 | 6.28 | ||||||||
| Granted | — | — | — | |||||||||
| Exercised | — | — | — | |||||||||
| Cancelled (unvested) | — | — | — | |||||||||
| Expired (vested) | — | — | — | |||||||||
| Balance outstanding, December 31, 2019 | 14,811,333 | $ | 0.62 | 5.33 | ||||||||
| Balance exercisable, December 31, 2019 | 14,811,333 | $ | 0.62 | 5.33 | ||||||||
As of December 31, 2019, 2018 and 2017, there was an aggregate of $0, $0, and $0 intrinsic value to the outstanding and exercisable options and warrants, respectively.
| -77- |
Warrants
In October 2017, the Company issued 1,000,000 fully vested Common Stock warrants to Landmark, in exchange for economic monetization and business mobilization services for the Company. The warrants are exercisable at any time from October 9, 2017 (initial exercise date) and on or prior to the close of business on the 10-year anniversary from the initial exercise date, October 8, 2027, at an exercise price of $0.25 per share. The warrants had a fair value of $0.15 per share and the Company has recognized $150,000 as consulting costs in the statement of operations during the fourth quarter ended December 31, 2017.
In November 2017, the Company issued 700,000 fully vested Common Stock warrants to a consultant for ongoing scientific and business consulting services. The warrants are exercisable at any time from November 14, 2017 (the grant date) for a period up to 10 years at an exercise price of $0.25 per share. The warrants had a fair value of $0.11 per share, determined using the Black-Scholes valuation model, and the Company has recognized $79,222 as consulting costs in the statement of operations during the further quarter ended December 31, 2017.
In April 2018, the Company issued an additional 1,000,000 fully vested Common Stock warrants to Landmark as final consideration paid upon completion of the 6-month Agreement. The Common Stock warrants are exercisable at any time from April 23, 2018 (initial exercise date) and on or prior to the close of business on the 10-year anniversary from the initial exercise date, April 22, 2028, at an exercise price of $0.25 per share. The warrants had a fair value of $0.08 per share, determined using the Black-Scholes valuation model. The Company recognized approximately $80,000, representing the aggregate fair value of the warrants as consulting expenses in the statement of operations during the second quarter ended June 30, 2018.
The
Company calculates the fair value of stock options using the Black-Scholes option-pricing model which approximates a binomial
lattice model. In determining the expected term,
The
following table summarizes the stock options and warrants that we granted during the year ended December
| Grant Date | Quantity Issued | Expected Life (Years) | Strike Price | Volatility | Dividend Yield | Risk-Free Interest Rate | Grant
Date Fair Value Per Warrant | Aggregate Fair Value | ||||||||||||||||||||||||
| 04/23/2018 | 1,000,000 | 10.0 | $ | 0.25 | 126.00 | % | 0 | % | 2.47 | % | $ | 0.08 | $ | 80,000 | ||||||||||||||||||
| 11/14/2017 | 700,000 | 10.0 | $ | 0.25 | 116.47 | % | 0 | % | 2.33 | % | $ | 0.11 | 79,222 | |||||||||||||||||||
| 10/09/2017 | 1,000,000 | 10.0 | $ | 0.25 | 115.00 | % | 0 | % | 2.47 | % | $ | 0.16 | 150,000 | |||||||||||||||||||
Note 11—Subsequent Events
Transactions with Shanxi Taxus Pharmaceuticals Co., Ltd.
On April 10, 2020, the Company entered into a Reaffirmation and Ratification Agreement with Shanxi . Shanxi has previously paid a subscription of $600,000 to acquire shares of our Common Stock or shares of capital stock in our Angionetics, Inc. subsidiary, confirming the application of the previously received $600,000 payment to the purchase of license rights to our Generx product candidate in greater China and have assigned our residual rights to Excellagen to Shanxi. The Company had previously entered into a strategic cooperation agreement and a subscription agreement with Shanxi. Mr. Jiayue Zhang, a member of our Board of Directors, is the Chairman and substantial stockholder of Shanxi. In connection with the entry into the agreements described, the Company terminated all prior agreements with Shanxi and entered into a mutual release of claims.
| -78- |
Amendment to Certificate of Incorporation and Amendment to Bylaws
On May 21, 2020, the Company amended their Certificate of Incorporation with the filing of a Certificate of Designation to establish the rights, privileges, and preferences of a new class of our preferred stock designated Series B Convertible Preferred Stock (“Series B Preferred Stock”). The Series B Preferred to have the following material terms and provisions:
Dividends. Each share of Series B Convertible Preferred Stock is entitled to receive dividends when, as, and if dividends are paid on shares of the Company’s Common Stock. Dividends are payable on each share Series B Convertible Preferred Stock on an “as-converted” basis, in the same amount and form as dividends actually paid on shares of our Common Stock. The Company has never paid dividends on shares of our common stock and the Company does not intend to do so for the foreseeable future.
Voting Rights. Each share of Series B Convertible Preferred Stock has the same voting rights as shares of Common Stock, on an “as-converted” basis, and votes on all matters with the Common Stock as a single class. In addition, the Series B Convertible Preferred Stock has voting rights that require the approval of a majority of the outstanding shares of Series B Convertible Preferred Stock for any action to: (1) alter or change adversely the powers, preferences or rights given to the shares of the Series B Convertible Preferred Stock or alter or amend its certificate of designation, (2) authorize or create any class of shares ranking as to dividends, redemption or distribution of assets upon liquidation senior to, or otherwise pari passu with, the shares of Series B Convertible Preferred Stock, (3) amend the Company’s Certificate of Incorporation or other charter documents in any manner that adversely affects any rights of the holders of our Series B Convertible Preferred Stock, (4) increase the number of authorized shares of our Series B Convertible Preferred Stock, or (5) enter into any agreement with respect to any of the foregoing.
Conversion. The shares of our Series B Convertible Preferred Stock are convertible at any time at the option of the holder into shares of Common Stock at a ratio determined by dividing the Stated Value of such share of Series B Preferred Stock by the conversion price of $0.0113 per share of Common Stock. Accordingly, each share of Series B Convertible Preferred Stock is initially convertible into 88.5 shares of Common Stock. The conversion price is subject to adjustment in the case of share splits, share dividends, combinations of shares and similar recapitalization transactions. In addition, if the Company sells shares of Common Stock or Common Stock equivalents at a price less than the current conversion price, the conversion price of the Series B Convertible Preferred Stock will be reduced to equal eighty percent (80%) of the price at which such Common Stock or Common Stock equivalents are sold.
Liquidation. The Series B Convertible Preferred Stock has a liquidation preference. Upon any liquidation, dissolution or winding up of our company, after payment or provision for payment of our debts and other liabilities and before any distribution or payment is made to the holders of our common stock or any junior securities, the holders of our Series B Convertible Preferred Stock will first be entitled to be paid an amount equal to $1.00 per share plus any other fees, liquidated damages or dividends then owing, before our remaining assets will be distributed among the holders of the other classes or series of shares of our capital stock in accordance with our Certificate of Incorporation.
| -79- |
Classified Board of Directors. On May 22, 2020, the Board amended the Company’s bylaws to eliminate the classified Board. Directors will serve one-year terms until the next annual meeting of stockholders or until their successors are duly elected and qualified.
Nostrum Financing
In May 2020, after the period covered by this report, we entered into a Preferred Stock Purchase Agreement with Nostrum, selling Nostrum 1,700,000 shares of our newly authorized Series B Convertible Preferred Stock in exchange for $1,700,000. Each share of Series B Convertible Preferred Stock is convertible into 88.4956 shares of Common, which are convertible into 150,442,478 shares of Common Stock on a fully diluted basis. As of March 31, 2021, Nostrum controlled approximately 75.2% of the voting interests of our Company.
In addition, Nostrum has purchased 220 shares of our Series A Convertible Preferred Stock from Sabby Master Healthcare Ltd., which are convertible into 19,469,026 shares of our Common Stock, and Nostrum has agreed to purchase up to 570 additional Series A Convertible Preferred Stock from Sabby. The Certificate of Designation of Preferences, Rights and Limitations of the Series A Convertible Preferred Stock restricts Nostrum from converting any Series A Preferred Stock if Nostrum would beneficially own a number of shares of Common Stock in excess of 9.99% of the shares of Common Stock then issued and outstanding. As a result of its ownership of the Series B Convertible Preferred Stock, Nostrum is currently limited in its entirety from converting any shares of Series A Convertible Preferred Stock into the Company’s Common Stock. The Series A Convertible Preferred Stock has no voting rights on general corporate matters, provided that the Series A Convertible Preferred Stock contain customary protective provisions.
The
Company will use the proceeds from the sale of the Series B Preferred Stock to fund working capital requirements in preparation
for conducting a Phase 3 clinical trial in the United States for
Conversion of Series A Convertible Preferred Stock
The Series B Convertible Preferred Stock financing resulted in a reset of the conversion price of our outstanding Series A Convertible Preferred Stock, such that the stated value of $1,000 for each share of Series A Convertible Preferred Stock is convertible into Common Stock at a price of $0.0113 per share of Common Stock. In a separate but concurrent transaction, when Nostrum acquired the 1,700,000 shares of Series B Convertible Preferred Stock, it also acquired 220 shares of Series A Convertible Preferred Stock from Sabby. Nostrum also agreed to purchase the remaining up to 570 shares of Series A Convertible Preferred Stock from Sabby within one year of the initial acquisition. Sabby retains the right prior to any such sale, to convert the Series A Convertible Preferred Stock. Since May 2020 to March 31, 2021, such holder has converted 397 shares of Series A Preferred Stock into 35,132,755 shares of our Common stock, that has increased our outstanding Common Stock to 49,622,154 shares as of March 31, 2021. As a result of these transactions, Nostrum currently controls approximately 75.2% of the voting interests of our Company.
Impact of Coronavirus Outbreak
On
January 30, 2020, the WHO announced the COVID-19 outbreak and the risks to the international community as the virus spreads globally
beyond its point of origin. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic, based on the rapid increase
in
The full impact of the COVID-19 outbreak continues to evolve as of the date of this report. As such, it is uncertain as to the full magnitude that the pandemic will have on the Company’s financial condition, liquidity, and future results of operations. Management is actively monitoring the impact of the global situation on its financial condition, liquidity, operations, suppliers, industry, and workforce. Given the daily evolution of the COVID-19 outbreak and the global responses to curb its spread, the Company is not able to estimate the effects of the COVID-19 outbreak on its results of operations, financial condition, or liquidity for fiscal year 2020 and 2021.
Office Lease
On July 8, 2020, the Company entered into a twenty-nine-month lease for approximately 3,039 square feet of office space in San Diego, California commencing on August 1, 2020. The monthly base rent is $6,685 and increases by three percent (3%) on each anniversary of the commencement date.
Resignation of Members of the Board of Directors
On
May 22, 2020, Andrew Leitch, John Wallace, Jiayue Zhang and Wei-Wei Zhang resigned as members of the Company’s Board of
Directors. The resignations were required under the terms of the Series B Preferred Stock Purchase Agreement. On May 22, 2020,
at the request of Nostrum, James Grainer and
| -80- |
Supplemental Quarterly Information (Unaudited)
The following tables set out our unaudited consolidated quarterly financial data, including summary balance sheet information and statement of operations for the quarterly periods during each of the years ended December 31, 2019, 2018 and 2017. The information has been prepared on a basis consistent with that of the audited consolidated financial statements included in this report. The Company’s management believes that all necessary adjustments, consisting of normal recurring accruals and adjustments, have been included to present fairly the quarterly financial data. Results of operations for any one period are not necessarily indicative of future results of operations.
The table below includes quarterly data:
| Year Ended December 31, 2019 | ||||||||||||||||||||
| 1st | 2nd | 3rd | 4th | Total | ||||||||||||||||
| Total current assets | $ | 18,065 | $ | 12,861 | $ | 54,021 | $ | 32,795 | $ | 32,795 | ||||||||||
| Property and equipment, net | $ | 44,873 | $ | 28,930 | $ | 12,988 | $ | 2,640 | 2,640 | |||||||||||
| Total assets | $ | 62,938 | $ | 41,791 | $ | 67,009 | $ | 35,435 | $ | 35,435 | ||||||||||
| Total current liabilities | $ | 5,944,935 | $ | 6,175,121 | $ | 6,230,401 | $ | 4,762,989 | $ | 4,762,989 | ||||||||||
| Total liabilities | $ | 5,944,935 | $ | 6,175,121 | $ | 6,350,796 | $ | 4,885,198 | $ | 4,885,198 | ||||||||||
| Total liabilities and stockholders’ deficit | $ | 62,938 | $ | 41,791 | $ | 67,009 | $ | 35,435 | $ | 35,435 | ||||||||||
| Research and development expense | $ | 63,379 | $ | 60,355 | $ | 61,443 | $ | 58,276 | $ | 243,453 | ||||||||||
| Selling, general and administrative expense | $ | 179,599 | $ | 180,473 | $ | 114,047 | $ | 119,430 | $ | 593,549 | ||||||||||
| Income (loss) from operations | $ | (242,978 | ) | $ | (240,828 | ) | $ | (175,490 | ) | $ | (177,706 | ) | $ | (837,002 | ) | |||||
| Other income (expenses) | $ | (10,129 | ) | $ | (10,504 | ) | $ | 25,033 | $ | 1,611,730 | $ | 1,616,130 | ||||||||
| Net Income (Loss) | $ | (253,107 | ) | $ | (251,332 | ) | $ | (150,457 | ) | $ | 1,434,024 | $ | 779,128 | |||||||
| Net Loss attributable to the controlling interest | $ | (226,369 | ) | $ | (226,369 | ) | $ | (130,667 | ) | $ | 1,450,080 | $ | 866,675 | |||||||
| Basic net income (loss) per share | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.01 | ) | $ | 0.10 | $ | 0.05 | |||||||
| Diluted net income (loss) per share | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.01 | ) | $ | 0.10 | $ | 0.05 | |||||||
| -81- |
| Year Ended December 31, 2018 | ||||||||||||||||||||
| 1st | 2nd | 3rd | 4th | Total | ||||||||||||||||
| Total current assets | $ | 12,708 | $ | 48,348 | $ | 199,718 | $ | 101,080 | $ | 101,080 | ||||||||||
| Property and equipment, net | $ | 108,075 | $ | 92,133 | $ | 76,757 | $ | 60,815 | $ | 60,815 | ||||||||||
| Total assets | $ | 120,783 | $ | 140,481 | $ | 276,475 | $ | 161,895 | $ | 161,895 | ||||||||||
| Total current liabilities | $ | 5,582,253 | $ | 5,861,727 | $ | 5,634,045 | $ | 5,790,786 | $ | 5,790,786 | ||||||||||
| Total liabilities | $ | 5,582,253 | $ | 5,861,727 | $ | 5,634,045 | $ | 5,790,786 | $ | 5,790,786 | ||||||||||
| Total liabilities and stockholders’ deficit | $ | 120,783 | $ | 140,481 | $ | 276,475 | $ | 161,895 | $ | 161,895 | ||||||||||
| Research and development expense | $ | 62,767 | $ | 63,049 | $ | 66,442 | $ | 63,136 | 255,394 | |||||||||||
| Selling, general and administrative expense | $ | 234,630 | $ | 267,629 | $ | 207,279 | $ | 198,298 | $ | 907,836 | ||||||||||
| Gain on sale of assets and technology | $ | - | - | (650,000 | ) | - | (650,000 | ) | ||||||||||||
| Income (loss) from operations | $ | (297,397 | ) | $ | (330,678 | ) | $ | 376,279 | $ | (261,434 | ) | $ | (513,230 | ) | ||||||
| Other income (expenses) | $ | (7,927 | ) | $ | (9,097 | ) | $ | (12,603 | ) | $ | (9,887 | ) | $ | 39,514 | ||||||
| Net Income (Loss) | $ | (305,324 | ) | $ | (339,775 | ) | $ | 363,676 | $ | (271,321 | ) | $ | (552,744 | ) | ||||||
| Net Loss attributable to the controlling interest | $ | (275,973 | ) | $ | (312,207 | ) | $ | 395,351 | $ | (242,052 | ) | $ | (434,881 | ) | ||||||
| Basic net income (loss) per share | $ | (0.02 | ) | $ | (0.02 | ) | $ | 0.03 | $ | (0.02 | ) | $ | (0.03 | ) | ||||||
| Diluted net income (loss) per share | $ | (0.02 | ) | $ | (0.02 | ) | $ | 0.03 | $ | (0.02 | ) | $ | (0.03 | ) | ||||||
| Year Ended December 31, 2017 | ||||||||||||||||||||
| 1st | 2nd | 3rd | 4th | Total | ||||||||||||||||
| Total current assets | $ | 215,571 | $ | 52,691 | $ | 18,664 | $ | 73,989 | $ | 73,989 | ||||||||||
| Other long-term assets | $ | 11,766 | $ | 11,766 | $ | 11,767 | $ | 11,767 | $ | 11,767 | ||||||||||
| Property and equipment, net | 146,591 | 155,902 | 139,959 | 124,017 | 124,017 | |||||||||||||||
| Total assets | $ | 373,928 | $ | 220,359 | $ | 170,390 | $ | 209,773 | $ | 209,773 | ||||||||||
| Total current liabilities | $ | 4,311,992 | $ | 4,600,648 | $ | 4,972,262 | $ | 5,355,655 | $ | 5,355,655 | ||||||||||
| Total liabilities | $ | 4,318,879 | $ | 4,624,785 | $ | 4,993,675 | $ | 5,365,920 | $ | 5,365,920 | ||||||||||
| Total liabilities and stockholders’ deficit | $ | 373,928 | $ | 220,359 | $ | 170,390 | $ | 209,773 | $ | 209,773 | ||||||||||
| Research and development expense | $ | 130,762 | $ | 66,182 | $ | 61,271 | $ | 86,761 | $ | 344,976 | ||||||||||
| Selling, general and administrative expense | $ | 522,565 | $ | 389,387 | $ | 351,173 | $ | 518,184 | $ | 1,781,309 | ||||||||||
| Gain on sale of assets and technology | - | - | - | (50,000 | ) | (50,000 | ) | |||||||||||||
| Income (loss) from operations | $ | (653,327 | ) | $ | (455,569 | ) | $ | (412,444 | ) | $ | (554,945 | ) | $ | (2,076,285 | ) | |||||
| Other income (expenses) | (1,956 | ) | $ | (4,709 | ) | $ | (6,414 | ) | $ | (7,140 | ) | $ | 20,219 | |||||||
| Net Income (Loss) | $ | (655,283 | ) | $ | (460,278 | ) | $ | (418,858 | ) | $ | (562,085 | ) | $ | (2,096,504 | ) | |||||
| Net Loss attributable to the controlling interest | $ | (580,123 | ) | $ | (417,493 | ) | $ | (375,322 | ) | $ | (521,204 | ) | $ | (1,894,142 | ) | |||||
| Basic net income (loss) per share | $ | (0.04 | ) | $ | (0.02 | ) | $ | (0.03 | ) | $ | (0.04 | ) | $ | (0.13 | ) | |||||
| Diluted net income (loss) per share | $ | (0.04 | ) | $ | (0.02 | ) | $ | (0.03 | ) | $ | (0.04 | ) | $ | (0.13 | ) | |||||
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Disclosure Controls and Procedures
We
maintain certain disclosure controls and procedures
| -82- |
Internal Controls Over Financial Reporting
We
maintain internal controls and procedures over financial reporting
Management,
including our principal executive officer and principal financial officer, do not expect that our internal controls will prevent
or detect all errors and all fraud. A control system, no matter how well designed and operated, can only provide reasonable, not
absolute, assurance that the objectives of the control system will be met. The design of a control system must reflect that there
are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations
in all control systems, no evaluation of internal
Under
the supervision, and with the participation of our management, including our
A
material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there
is a reasonable possibility that a material misstatement of our annual or interim consolidated financial statements will not be
prevented or detected and corrected on a timely basis. At the year ended December
| ● |
Our management does not believe that the material weaknesses in internal controls has resulted in any inaccuracy or misstatement in the financial statements included in this report. We plan to remediate these material weaknesses by hiring additional qualified accounting personnel when the Company has the financial resources to support those expenses.
This
annual report does not include an attestation report of our registered public accounting firm regarding internal control over
financial reporting. Management
Changes in Internal Control Over Financial Reporting
There
were no material changes in and 2017.
| ITEM 9B. | OTHER INFORMATION |
None.
| -83- |
Directors and Executive Officers
The
following table sets forth information regarding our current directors and executive officers and their ages as of March
|
|
| |||||||||||||||||||||||||||||||||||||||
Christopher
J. Reinhard is co-founder of the Company and has served as a director and the Chief Executive Officer and President of
Ronald
J. Shebuski, PhD. currently serves as the Company’s Chief Scientific Officer. Dr. Shebuski has over 25 years
of experience in the pharmaceutical industry. From 2016 to 2020 he served as the Vice President of Research and Development for
InspiRx, Inc. From 1998 to 2016 he established Cardiovascular Research Consulting, LLC, an advisory firm serving small, mid, and
large pharma companies in many aspects of drug discovery. From 1990 to 1998 he served as the Director of Cardiovascular Therapeutics
at Pharmacia & Upjohn, where he managed the team responsible for FDA approval of the Class III anti-arrhythmic agent, Corvert®.
Prior to Pharmacia & Upjohn, Dr. Shebuski was a Senior Scientist responsible for leading cardiovascular drug discovery teams
at Merck Research Laboratories and Smith Kline & French in the Philadelphia area. His primary field of research has focused
on thrombosis and the development of effective new thrombolytic, anti-platelet and anti-coagulant therapies. At Merck, Dr. Shebuski
led the in vivo discovery and development effort which culminated in the identification and eventual FDA approval of the anti-platelet
GPIIb/IIIa antagonist, Aggrastat®. He also led development teams in identification of novel factor Xa and P- selectin antagonists
at Merck and Pharmacia & Upjohn, respectively. Dr. Shebuski received his B.S. Degree in Microbiology from the University of
Wisconsin at Madison and Ph.D. Degree in Pharmacology from the University of Minnesota Medical School. Further, Dr. Shebuski is
expert in FDA regulations and has an ongoing appointment (since 2009) with FDA as a Special Government Employee (SGE) in the CardioRenal
Division of CDER (Center for Drug Evaluation and Research). Lois
A. Chandler, PhD. serves as the Company’s Chief Operating Officer. Dr. Chandler has 20 years of professional experience
in biopharmaceutical design and clinical development, Chemistry, Manufacturing and Controls, Quality Control and Regulatory Affairs.
Dr. Chandler joined Cardium Therapeutics in 2004, with the acquisition of Tissue Repair Company, and managed internal activities
leading to FDA 510(k) clearance of the Excellagen advanced wound care product. Dr, Chandler is currently the Company’s authorized
representative with FDA and has served as lead author of recent Generx FDA submissions including transfer of the Murray
H. Hutchison has Kaushik
K. Vyas joined our Board of Directors in May 2020. He currently serves as the Chief Executive Officer Role
of the Board of Directors Our
Board of Directors oversees the CEO and other senior management in the competent and ethical operation of our business on a day-to-day
Our
Board of Directors is currently comprised of four members. Our Board of Directors has determined that two of our directors—Messrs.
Hutchison and Our
key governance documents, including our Corporate Governance Guidelines, are available at www.genebiotherpautics.com. The Board
had no meetings in Board
Leadership Structure Our
Board leadership structure currently separates the role of the Chairman and the Chief Executive Officer. Mr. Reinhard had served
as our Chairman of the Board since our initial founding. Following the investment by Nostrum in May 2020, we revised our Board
leadership structure to appoint Mr. Grainer as our Chief Financial Officer, and as a long-time employee of Nostrum Pharmaceuticals
LLC, he was appointed as our Chairman of the Board. Our
Board believes its current leadership structure best serves the objectives of the Board’s oversight of management, the Board’s
ability to carry out its roles and responsibilities on behalf of Gene Biotherapeutic’s stockholders and Gene Biotherapeutic’s
overall corporate governance. The Board also believes that separation of the Chairman and Chief Executive Officer roles allows
our Chief Executive Officer to focus his time and energy on operating and managing Gene Biotherapeutics, while leveraging our
Chairman’s experience and perspectives. The Board periodically reviews its leadership structure to determine whether it
continues to best serve Gene Biotherapeutics and its stockholders. Board
Oversight or Risk Management. The
Board believes that evaluating the executive team’s management of the various risks confronting our business is one of its
most important areas of oversight. In carrying out this critical responsibility. The Board has designated the Audit Committee
with primary responsibility for overseeing enterprise risk management. In accordance with this responsibility, the Audit Committee
monitors our significant business risks, including financial; operational; privacy; data security; business continuity; tax; legal
and regulatory compliance; and reputational risks. The Audit Committee reviews the steps management has taken to monitor and mitigate
these risks. The other Board committees also consider risks within their areas of responsibility and apprise the Board of significant
risks and management’s response to those risks. The Nominating Committee reviews legal and regulatory compliance risks as
they relate to Apple’s corporate governance structure and processes, and the Compensation Committee reviews risks related
to compensation matters. While the Board and its committees oversee risk management strategy, management is responsible for implementing
and supervising day-to-day risk management processes and reporting to the Board and its committees. Board
Committees Our
Board of Directors has a separately designated standing Audit Committee, Remuneration Committee and Nomination Committee. Each
committee operates under a written charter adopted by the Board, which is available on our website at www.genebiotherapeutics.com.
We maintained our board committees consistent with the corporate governance and director independence standards required by Nasdaq
throughout the periods covered by this report. Following the Nostrum transaction in May 2020, a number of our directors resigned,
Compensation
Committee Nominating
Committee
The
Audit Committee met one time in 2017 and had no meetings in 2018 and 2019. Compensation
Committee. Our Compensation Committee is
Nominating
Committee. Our Nominating and Corporate Governance Committee consists of four members. Our Board of Directors has determined
that each member of the committee is “independent” as that term is defined in the applicable SEC and Nasdaq rules.
Our Nominating and Corporate Governance Committee is authorized to: Code
of Ethics We
have adopted a Code of Ethics that applies to all Attendance
at Annual Meetings In
recognition that it may not be possible or practicable, in light of other business commitments of the Company’s directors,
to attend the Company’s annual meetings of stockholders, the members of the Board of Directors are invited, but not required,
Stockholder
Communications with Directors Our
Board of Directors has adopted a Stockholder Communications Policy to provide a process by which our stockholders may communicate
with our Board of Directors. Under the policy, stockholders may communicate with our Board of Directors as a whole, with the independent
directors, with all members of a committee of our Board of Directors, or with a particular director. Stockholders wishing to communicate
directly with our Board of Directors may do so by mail addressed to the Company at Report
of the Audit Committee As
of the date of this report, the Audit Committee consisted of four members: James L. Grainer, who serves as the Chair of the Committee,
Christopher Reinhard, Murray Hutchison, and Kaushik K. Vyas. The
Audit Committee oversees the financial reporting process on behalf of the Board of Directors. The Audit Committee has the duties
and powers described in its written charter adopted by the Board. The Audit Committee is responsible for the appointment, compensation,
retention, and oversight of the work performed by Apple’s independent registered public accounting firm, Marcum LLP. In
fulfilling its oversight responsibility, the Audit Committee carefully reviews the policies and procedures for the engagement
of the independent registered public accounting firm, including the scope of the audit, audit fees, auditor independence matters,
performance of the independent auditors, and the extent to which the independent registered public accounting firm may be retained
to perform non-audit services. The
Audit Committee has reviewed and discussed the audited financial statements for the years ended December 31, 2019, 2018 and 2017
with Gene Biotherapeutics’ management and Marcum LLP. The Audit Committee has also discussed with Marcum LLP the matters
required to be discussed by the applicable requirements of the Public Company Accounting Oversight Board (“PCAOB”)
and the SEC. The Audit Committee also has received and reviewed the written disclosures and the letter from Marcum LLP required
by applicable requirements of the PCAOB regarding Marcum LLP’s communications with the Audit Committee concerning independence
and has discussed with Marcum LLP its independence. Based
on the reviews and discussions referred to above, the Audit Committee recommended to the Board that the financial statements referred
to above be included in The Company’s Annual Report on Form 10-K for the year ended December 31, 2019 for filing with the
SEC. It
is not the duty of the Audit Committee to plan or conduct audits or to determine that the Company’s financial statements
are complete and accurate and are prepared in accordance with generally accepted accounting principles; that is the responsibility
of Submitted
by the members of the Audit Committee
James
L. Grainer, Chairman Christopher
J. Reinhard
Murray
H. Hutchison The
following table shows the compensation earned by, or paid or awarded to, each person who served as our chief executive officer
or chief financial officer during the Summary
Compensation Table 2019, 2018 and 2017 Name
and Principal
Position Nonqualified ($) Narrative
Disclosure to 2019 Summary Compensation Table in place with Mr. Reinhard or any other executive officer or employee of the Company. Outstanding
Equity Awards at Fiscal Year-End The
following table provides certain information about unexercised option awards and unvested restricted stock awards held by our
named executive officers as of December Number
of Securities Underlying Unexercised (#) Exercisable Number
of Securities Underlying Unexercised Warrants
– (#)
Un- Exercisable Equity Incentive Plan Awards: Number
of Securities Underlying Unexercised, Unearned Options
(#) Warrant Exercise Price ($) Warrant Expiration Date Number of Shares or Units
of Stock That Have Not Vested (#) Market Value of Shares or Units of
Stock That Have Not Vested ($) Equity Incentive Plan Awards: Number of Unearned Shares, Units
or Other Rights That Have
Not (#) Equity Incentive Plan Awards: Market or
Payout Value
of Unearned Shares, Units
or Other Rights That Have
Not Vested (#) During
the Due
to financial hardship, we did not pay our non-employee directors any compensation, cash, or equity, for their services as directors
during the
The
following table sets forth information on the beneficial ownership of our common stock as of -84- -85-
James
L. Grainer (Chair)
James
L. Grainer (Chair)
Christopher
J. Reinhard
Christopher
J. Reinhard
Christopher
J. Reinhard
Murray
H. Hutchison
Murray
H. Hutchison
Kaushik
K. Vyas
Kaushik
K. Vyas
Kaushik
K. Vyas
●
approve and retain the independent
●
review the proposed scope and results of the
audit;
●
review and pre-approve audit and non-audit fees
and services;
●
review accounting and financial controls with
the independent auditors and our financial and accounting staff;
●
review and approve transactions between us and
our directors, officers, and affiliates;
●
recognize and prevent prohibited non-audit services;
●
establish procedures for complaints received
by us regarding accounting matters; and
●
oversee internal audit functions, if any. -86-
●
review and determine the compensation arrangements
for management;
●
establish and review general compensation policies
with the objective to attract and retain superior talent, to reward individual performance and to achieve our financial goals;
●
administer our stock incentive and purchase
plans;
●
oversee the evaluation of the board of directors
and management; and
●
review the independence of any compensation
advisers engaged by the compensation committee.
The Compensation Committee did not
meet in 2019, 2018 and 2017.
●
identify, evaluate,
and make recommendations to our board of directors regarding prospective director nominees;
●
evaluate and make
recommendations to our board of directors regarding the compensation of our board of directors and its committees;
●
oversee the evaluation
of our board of directors and its committees;
●
review developments
in corporate governance practices;
●
evaluate the adequacy
of our corporate governance practices and reporting; and
●
develop, periodically
review, and make recommendations to our board of directors regarding corporate governance guidelines and matters.
The
Nominating Committee did not meet in 2019, 2018 -87-
Year
Salary
($)
Bonus
($)
Stock
Awards
($)
Option
Awards
($)
Non-Equity
Incentive Plan
Compensation
($)
Deferred
Compensation
Earnings
All
Other
Compensation
($)
Total
($)
Christopher J. Reinhard
2019
239,000
—
—
—
—
—
—
239,000
Chief
Executive Officer
2018
235,000
—
—
—
—
—
—
235,000
2017
234,100
—
—
—
—
—
—
234,100
Warrants
Stock
Awards
Name
Warrants –
Vested
Christopher J. Reinhard
2,438,000
—
—
$ 0.80
02/28/2024
—
—
—
—
1,333,333
—
—
$ 0.60
03/23/2025
—
—
—
— -89-
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS ’s spouse. Except as otherwise indicated, the business address for each beneficial owner is 11230 Sorrento
Valley Road, Suite 220, San Diego, California 92121.
| Name of Beneficial Owner | Shares of Common Stock Beneficially Owned |
Percent of Common Stock Outstanding |
||||||
| Nostrum Pharmaceuticals, LLC | 150,442,478 | 1 | 75.2 | % | ||||
| Sabby Management LLC | 4,839,784 | 2 | 9.9 | % | ||||
| Jiayue Zhang | 3,848,953 | 3 | 8.7 | % | ||||
| Christopher J. Reinhard | 3,911,929 | 4 | 8.2 | % | ||||
| James L. Grainer | — | — | % | |||||
| Murray H. Hutchison | 388,889 | 5 | 0.9 | % | ||||
| Kaushik K. Vyas | — | — | % | |||||
| All directors and executive officers as a group (6 persons) | 4,300.818 | 6 | 8.9 | % | ||||
|
|
Includes
|
| -90- |
|
|
Includes |
Since the period beginning January 1, 2017, in addition to the transactions described under “Executive Officer Compensation” and “Director Compensation” above, we entered the following related party transactions:
| ● | Our Chief Executive
Officer, Christopher Reinhard, a co-founder of a predecessor company, and founder of Gene Biotherapeutics has historically
provided his own personal interest-free loans to support the Company’s on-going operations. The outstanding balance
due and payable to Mr. Reinhard as of December 31,
these transactions and is the Chairman and a substantial stockholder of Shanxi. In
connection with the Shanxi License Agreement we applied a $600,000 subscription to payment of a license fee granted Shanxi
(a) a non-exclusive right to manufacture Generx products in China, and (b) an exclusive right to market and sell Generx products
in Singapore, Macau, Hong Kong, Taiwan, any other municipality other than mainland China where Chinese (Mandarin or Cantonese)
is the common language, the Russian Federation and the CIS (i.e., Armenia, Azerbaijan, Belarus, Kazakhstan, Kyrgyzstan,
Moldova, Tajikistan, Turkmenistan, and Uzbekistan). The Shanxi License Agreement provides for a royalty ranging from 5% up
to 10% based on the level of annual net sales of the Generx product sold by Shanxi in the licensed territory. |
| -91- |
On
|
The
following table shows the
| 2019 | 2018 | 2017 | ||||||||||
| Audit Fees1 | - | $ | - | $ | 151,210 | |||||||
| Audit Related Fees2 | - | - | - | |||||||||
| Tax Fees3 | - | - | - | |||||||||
| All Other Fees4 | - | - | - | |||||||||
| Total | $ | - | $ | - | $ | 151,210 | ||||||
| 1 | Audit
fees related to professional services rendered | |
| 2 | Audit
related fees relate to professional services that are reasonably related to the performance of the audit or review of | |
| 3 | Tax fees relate to professional services rendered in connection with tax compliance and preparation related to tax returns and tax audits, as well as for tax consulting and planning services. | |
| 4 | All
Other fees relate to professional services not included in the categories above, |
Pre-Approval Policies and Procedures
The following documents are filed as part of this report:
(1)
Financial Statements. The financial statements listed below are included under Item
Consolidated
Balance Sheets as of December |
| ||
| ● | Consolidated
Statements of Operations for the years ended December |
| ||
| ● | Consolidated
Statements of Stockholders |
| ||
| ● | Consolidated
Statements of Cash Flows for the years ended December |
| ||
| ● | Notes to Consolidated Financial Statements. |
(2)
Financial Statement Schedules.
(3)
EXHIBIT INDEX
Exhibit |
Description |
Incorporated by Reference To | ||
| 3.1 | Exhibit 3(i) of our Registration Statement on Form SB-2 (File No. 333-131104), filed with the SEC on January 18, 2006. | |||
| 3.2 | Certificate of Ownership and Merger of the registrant effective as of March 14, 2014 | Exhibit 3.1 of our Current report on Form 8-K, filed with the SEC on March 18, 2014. | ||
| 3.2 | Amended and Restated Bylaws of the registrant effective as of January 12, 2006 | Exhibit 3(ii) of our Registration Statement on Form SB-2 (File No. 333-131104), filed with the SEC on January 18, 2006. | ||
| 3.3 | Certificate of Designation for Series A Convertible Preferred Stock | Exhibit 3.1 of our Current Report on Form 8-K, filed with the SEC on April 5, 2013. | ||
| 3.4 | Certificate of Designation for Series B Convertible Preferred Stock | Exhibit 3.1 of our Current Report on Form 8-K, filed with the SEC on May 28, 2020. | ||
| 4.1 | Description of Securities of the registrant | Filed herewith. | ||
| 4.2 | Form of Warrant Agreement issued to directors and officers in February 2014. | Exhibit 4.1 of our Form 10-Q, filed with the SEC on May 15, 2014. | ||
| 10.1 | Technology Transfer Agreement effective as of October 13, 2005 among the registrant and Schering AG, Berlex, Inc. and Collateral Therapeutics, Inc. | Exhibit 10.5 of our Current Report on Form 8-K, filed with the SEC on October 26, 2005. | ||
| 10.2 | Securities Purchase Agreement dated April 4, 2013 between the registrant and Sabby Master Volatility Fund for the purchase of Series A Convertible Preferred Stock. | Exhibit 10.1 of our Current Report on Form 8-K, filed with the SEC on April 5, 2013. | ||
| 10.3 | Share Purchase Agreement dated June 7, 2016 among the registrant, Angionetics Inc. and Pineworld Capital Limited | Exhibit 10.1 of our Current Report on Form 8-K filed with the SEC on July 11, 2016. | ||
| -93- |
| Exhibit Number |
Description | Incorporated by Reference To | ||
| 10.4 | Distribution and License Agreement dated June 7, 2016 between Angionetics Inc. and Pineworld Capital Limited | Exhibit 10.1 of our Current Report on Form 8-K filed with the SEC on July 11, 2016. | ||
| 10.5 | Asset Purchase Agreement dated July 15, 2018 between Activation Therapeutics, Inc. and Olaregen Therapeutix, Inc. for the sale of Excellagen. | Filed herewith. | ||
| 10.6 | Reaffirmation and Ratification Agreement dated April 10, 2020 between the registrant and Shanxi Taxus Pharmaceuticals Co. Ltd. | Exhibit 10.2 of our Current Report on Form 8-K filed with the SEC on May 28, 2020. | ||
| 10.7 | Distribution and License Agreement Dated April 10, 2020 between Angionetics, Inc. and Shanxi Taxus Pharmaceuticals Co., Ltd. | Exhibit 10.3 of our Current Report on Form 8-K filed with the SEC on May 28, 2020. | ||
| 10.8 | Amendment No. 1 to Distribution and License Agreement dated April 14, 2020 between Angionetics, Inc. and Shanxi Taxus Pharmaceuticals Co., Ltd. | Exhibit 10.4 of our Current Report on Form 8-K filed with the SEC on May 28, 2020. | ||
| 10.9 | License and Patent Assignment Agreement dated April 10, 2020 between Activation Therapeutics, Inc. and Shanxi Taxus Pharmaceuticals Co., Ltd. | Exhibit 10.5 of our Current Report on Form 8-K filed with the SEC on May 28, 2020 | ||
| 10.10 | Preferred Stock Purchase Agreement dated May 22, 2020 between the registrant and Nostrum Pharmaceuticals, LLC, for the purchase of Series B Convertible Preferred Stock. | Exhibit 10.1 of our Current Report on Form 8-K filed with the SEC on May 28, 2020 | ||
| 21.1 | Subsidiaries of the registrant | Filed herewith | ||
| 24.1 | Power of Attorney | Filed herewith | ||
| 31.1 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Included on signature page of this report | ||
| 31.2 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Filed herewith | ||
| 32 | Section 1350 Certification of Chief Executive Officer and Chief Financial Officer | Filed herewith | ||
| 101 | Inline XBRL document Set for the consolidated financial statements and accompanying notes in Part II, Item 8 “Financial Statements and Supplemental Data” of this Annual Report on Form 10-K. | Filed herewith | ||
| ITEM 16. | FORM 10-K SUMMARY |
None
| -94- |
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934 Date:
April
Christopher
J. Reinhard, KNOW
ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby severally constitutes and appoints Christopher
J. Reinhard Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf
of Signature Title Date
GENE
BIOTHERAPEUTICS INC.
Chief Executive Officer (Principal Executive
April 23,
2021
Christopher J.
Reinhard
Officer) and Director
/S/
James L. Grainer
Chief Financial Officer (Principal Accounting
April 23,
2021
James L. Grainer
Officer) and Chairman of the Board of Directors
Director
Murray H. Hutchison
Director
Kaushik K. Vyas
-95-
Exhibit 4.1
Description of the Registrant’s Securities
The following descriptions of our common stock, our preferred stock and certain provisions of our certificate of incorporation and bylaws are summaries and are qualified by reference to the complete copies of our certificate of incorporation and bylaws with are exhibits to this report.
Gene Therapeutics, Inc. has one class of securities registered under Section 12 of the Securities Exchange Act of 1934, as amended, our common stock, par value $0.0001 per share.
Common Stock
Authorized Share Capital. Our certificate of incorporation authorizes us to issue up to 200,000,000 shares of common stock.
Voting Rights. The holders of our common stock are entitled to one vote per share on all matters submitted to a vote of our stockholders. There is no cumulative voting with respect to the election of directors, with the result that directors are elected by a plurality of the votes cast. There is no classification of the board of directors.
Dividend Rights. The holders of outstanding shares of common stock are entitled to receive ratably any dividends declared by our board of directors out of assets legally available therefor, subject to preferences that may be applicable to any preferred stock outstanding at the time.
Liquidation Preferences. In the event that we liquidate, dissolve or wind up, holders of our common stock are entitled to share ratably in all assets remaining after payment of liabilities and the liquidation preference of any then outstanding shares of preferred stock.
Other Rights. Holders of common stock have no preemptive or conversion rights or other subscription rights. There are no redemption or sinking fund provisions applicable to the common stock. All outstanding shares of common stock are fully paid and non-assessable.
Preferred Stock
Gene Therapeutics, Inc. has outstanding preferred stock. The preferred stock is not registered under Section 12 of the Securities Exchange Act of 1934, as amended. However, rights evidenced by the preferred stock may limit or qualify the rights of the holders of our common stock and we are including information on our preferred stock so investors may understand those limitations and qualifications.
Authorized Share Capital. Our certificate of incorporation authorizes us to issue up to 40,000,000 shares of preferred stock with rights, preferences and privileges of which may be designated from time to time by our board of directors. Our board of directors has authorized to series of preferred stock, our Series A Convertible Preferred Stock and Series B Convertible Preferred Stock.
| -1- |
Our board of directors may, without further action by our stockholders, fix the rights, preferences, privileges and restrictions of up to an aggregate of 38,295,988 shares of preferred stock in one or more series and authorize their issuance, subject to the approval rights of the holders of Series A Convertible Preferred Stock and Series B Preferred Stock described below. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of our common stock or outstanding preferred stock. The issuance of our preferred stock could adversely affect the voting power of holders of our common stock and the likelihood that such holders will receive dividend payments and payments upon liquidation. In addition, the issuance of preferred stock could have the effect of delaying, deferring or preventing a change of control or other corporate action.
Series A Convertible Preferred Stock
Our board of directors has designated a class of 4,012 shares of our preferred stock as “Series A Convertible Preferred Stock.” All 4,012 shares of Series B Preferred Stock have been issued and as of December 31, 2019 790 shares remained outstanding, with the following rights, privileges and preferences:
Voting Rights. Except as required by law, holders of the shares of our Series A Convertible Preferred Stock will not have rights to vote on any matters, questions or proceedings, including the election of directors. However, as long as any shares of our Series A Convertible Preferred Stock are outstanding, we cannot, without the affirmative vote of the holders of a majority of the then outstanding shares of our Series A Convertible Preferred Stock, (1) alter or change adversely the powers, preferences or rights given to the shares of our Series A Convertible Preferred Stock or alter or amend its certificate of designation, (2) authorize or create any class of shares ranking as to dividends, redemption or distribution of assets upon liquidation senior to, or otherwise pari passu with, the shares of our Series A Convertible Preferred Stock, (3) amend our Certificate of Incorporation or other charter documents in any manner that adversely affects any rights of the holders of our Series A Convertible Preferred Stock, (4) increase the number of authorized shares of our Series A Convertible Preferred Stock, or (5) enter into any agreement with respect to any of the foregoing. As long as any shares of Series A Convertible Preferred Stock are outstanding, unless the holders of more than two-thirds of the outstanding Series A Convertible Preferred Stock approve, we have agreed not to, directly or indirectly (1) incur any indebtedness other than Permitted Indebtedness (as defined in the certificate of incorporation), (2) incur any liens other than Permitted Liens, (3) amend our certificate of incorporation in a manner that adversely affects the rights of any holder of Series A Convertible Preferred Stock, (4) repurchase or redeem shares of our outstanding common stock or common stock equivalents, (5) pay any dividends on our common stock, or (6) enter into any related party transactions, except for arm’s-length transactions that are expressly approved by a majority of the disinterested directors of our board of directors.
Dividends. Each share of our Series A Convertible Preferred Stock is entitled to receive dividends when, as, and if dividends are paid on shares of our common stock. Dividends are payable on each share Series A Convertible Preferred Stock on an “as-converted” basis, in the same amount and form as dividends actually paid on shares of our common stock.
| -2- |
Liquidation Preferences. In the event that we liquidate, dissolve or wind up, after payment or provision for payment of our debts and other liabilities and before any distribution or payment is made to the holders of our common stock or any junior securities, the holders of our Series A Convertible Preferred Stock will first be entitled to be paid an amount equal to $1,000 per share plus any other fees, liquidated damages or dividends then owing, before our remaining assets will be distributed among the holders of the other classes or series of shares of our capital stock in accordance with our certificate of incorporation.
Conversion. Subject to certain ownership limitations as described below, the shares of our Series A Convertible Preferred Stock are convertible at any time at the option of the holder into shares of our common stock at a conversion ratio determined by dividing the stated value of the shares of our Series A Convertible Preferred Stock (or $1,000) by a conversion price of $0.0113 per share. The conversion price is subject to adjustment in the case of share splits, share dividends, combinations of shares and similar recapitalization transactions. We have the right to force conversion of the Series A Preferred Stock into common stock; provided that during a period of 30 consecutive trading days the VWAP for each of any 25 trading days during period exceeds $0.60 (subject to adjustment for forward and reverse stock splits and the like) and the dollar trading volume for each trading day during such period exceeds $2,000,000 per Trading Day, and we meet certain other “Equity Conditions” described in our certificate of incorporation. A holder of our Series A Convertible Preferred Stock will not have the right to convert, and we will not have the right to force such holder to convert, any portion of its shares of our Series A Convertible Preferred Stock if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number shares of our common stock outstanding immediately after giving effect to its conversion.
Series B Preferred Stock
Our board of directors has designated a class of 1,700,000 shares of our preferred stock as “Series B Convertible Preferred Stock.” All 1,700,000 shares of Series B Preferred Stock have been issued and are outstanding, with the following rights, privileges and preferences:
Voting Rights. Each share of our Series B Convertible Preferred Stock has the same voting rights as shares of our common stock, on an “as-converted” basis, and votes on all matters with the common stock as a single class. In addition, the Series B Convertible Preferred Stock has voting rights that require the approval of a majority of the outstanding shares of Series B Convertible Preferred Stock for any action to: (1) alter or change adversely the powers, preferences or rights given to the shares of our Series B Convertible Preferred Stock or alter or amend its certificate of designation, (2) authorize or create any class of shares ranking as to dividends, redemption or distribution of assets upon liquidation senior to, or otherwise pari passu with, the shares of our Series B Convertible Preferred Stock, (3) amend our Certificate of Incorporation or other charter documents in any manner that adversely affects any rights of the holders of our Series B Convertible Preferred Stock, (4) increase the number of authorized shares of our Series B Convertible Preferred Stock, or (5) enter into any agreement with respect to any of the foregoing.
Dividends. Each share of our Series B Convertible Preferred Stock is entitled to receive dividends when, as, and if dividends are paid on shares of our common stock. Dividends are payable on each share Series B Convertible Preferred Stock on an “as-converted” basis, in the same amount and form as dividends actually paid on shares of our common stock.
| -3- |
Conversion. The shares of our Series B Convertible Preferred Stock are convertible at any time at the option of the holder into shares of our common stock at a ratio determined by dividing the stated value of $1.00 per such share of Series B Preferred Stock by the conversion price of $0.0113 per share of common stock. Accordingly, each share of our Series B Convertible Preferred Stock is initially convertible into 88.5 shares of our common stock. The conversion price is subject to adjustment in the case of share splits, share dividends, combinations of shares and similar recapitalization transactions. In addition, if we sell shares of Common Stock or Common Stock equivalents at a price less than the current conversion price, the conversion price of the Series B Convertible Preferred Stock will be reduced to equal eighty percent (80%) of the price at which such common stock or common stock equivalents are sold.
Liquidation. The Series B Convertible Preferred Stock has a liquidation preference. Upon any liquidation, dissolution or winding up of our company, after payment or provision for payment of our debts and other liabilities and before any distribution or payment is made to the holders of our common stock or any junior securities, the holders of our Series B Convertible Preferred Stock will first be entitled to be paid an amount equal to $1.00 per share plus any other fees, liquidated damages or dividends then owing, before our remaining assets will be distributed among the holders of the other classes or series of shares of our capital stock in accordance with our Certificate of Incorporation.
Anti-Takeover Provisions
Certificate of Incorporation and Bylaws
Our certificate of incorporation and bylaws provide that annual and special meetings of stockholders may be called only the majority of our whole board of directors and not by the shareholders or any other person. Our bylaws provide for advance notice requirements for shareholder nominations for director.
The foregoing provisions will make it more difficult for our existing stockholders to replace our board of directors as well as for another party to obtain control of us by replacing our board of directors. Since our board of directors has the power to retain and discharge our officers, these provisions could also make it more difficult for existing stockholders or another party to effect a change in management. In addition, the authorization of undesignated preferred stock makes it possible for our board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change our control.
These provisions are intended to enhance the likelihood of continued stability in the composition of our board of directors and its policies and to discourage certain types of transactions that may involve an actual or threatened acquisition of us. These provisions are also designed to reduce our vulnerability to an unsolicited acquisition proposal and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and may have the effect of deterring hostile takeovers or delaying changes in our control or management. As a consequence, these provisions also may inhibit fluctuations in the market price of our stock that could result from actual or rumored takeover attempts.
| -4- |
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| ● | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; | |
| ● | upon closing of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned by (i) persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| ● | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines “business combination” to include the following:
| ● | any merger or consolidation involving the corporation and the interested stockholder; | |
| ● | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; | |
| ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; | |
| ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or | |
| ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
| -5- |
Limitations of Liability and Indemnification
Our certificate of incorporation provides that we will indemnify our directors and officers, and may indemnify our employees and other agents, to the fullest extent permitted by the Delaware General Corporation Law. However, Delaware law prohibits our amended and restated certificate of incorporation from limiting the liability of our directors for the following:
| ● | any breach of a director’s duty of loyalty to us or to our stockholders; | |
| ● | acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law; | |
| ● | unlawful payment of dividends or unlawful stock repurchases or redemptions; and | |
| ● | any transaction from which a director derived an improper personal benefit. |
If Delaware law is amended to authorize corporate action further eliminating or limiting the personal liability of a director, then the liability of our directors will be eliminated or limited to the fullest extent permitted by Delaware law, as so amended. Our certificate of incorporation does not eliminate a director’s duty of care and, in appropriate circumstances, equitable remedies, such as injunctive or other forms of non-monetary relief, remain available under Delaware law. It also does not affect a director’s responsibilities under any other laws, such as the federal securities laws or other state or federal laws. Under Delaware law and our bylaws, we are empowered to enter into indemnification agreements with our directors, officers, employees and other agents and to purchase insurance on behalf of any person whom we are required or permitted to indemnify.
The limitation of liability and indemnification provisions in our certificate of incorporation may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might benefit us and our stockholders. A stockholder’s investment may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions. Insofar as indemnification for liabilities arising under the Securities Act, may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act, and is, therefore, unenforceable. There is no pending litigation or proceeding naming any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director or officer.
| -6- |
Exhibit 10.5
ASSET PURCHASE AGREEMENT
BY AND BETWEEN
ACTIVATION THERAPEUTICS INC.
as Seller,
AND
OLAREGEN THERAPEUTIX INC.
as Buyer,
Dated as of July 15, 2018
TABLE OF CONTENTS
| ARTICLE I PURCHASE AND SALE OF ASSETS | 2 | |
| 1.1 | Purchase and Sale of Assets | 2 |
| 1.2 | Assumption of Liabilities & Reimbursement of Credit | 3 |
| 1.3 | Closing | 3 |
| 1.4 | Payment | 3 |
| 1.5 | Transfer Documents | 3 |
| 1.6 | Further Assurances | 4 |
| 1.7 | Transfer Taxes | 4 |
| ARTICLE II purchase price | 4 | |
| 2.1 | Purchase Price | 4 |
| ARTICLE III REPPRESENTATIONS AND WARRANTIES OF THE SELLER | 5 | |
| 3.1 | Organization, Good Standing and Qualification of the Seller | 5 |
| 3.2 | Authorization; Binding Obligation | 5 |
| 3.3 | Consents and Approvals | 5 |
| 3.4 | No Violation | 5 |
| 3.5 | Legal Proceedings | 6 |
| 3.6 | Title to Assets. | 6 |
| 3.7 | Intellectual Property. | 7 |
| 3.8 | FDA Compliance; Compliance with Healthcare Laws. | 7 |
| ARTICLE IV REPRESENTATIONS AND WARRANTIES OF THE BUYER | 9 | |
| 4.1 | Organization and Good Standing | 9 |
| 4.2 | Authorization; Binding Obligation | 9 |
| 4.3 | Consents and Approvals | 9 |
| 4.4 | No Violation | 9 |
| 4.5 | Legal Proceedings | 9 |
| 4.6 | Audit Rights | 10 |
| 4.7 | Seniority of the Total Additional Consideration | 10 |
| 4.8 | Accelerated Payment of Total Additional Consideration | 10 |
| 4.9 | Financial Ability | 10 |
| ARTICLE V COVENANTS | 10 | |
| 5.1 | Conduct of Business Pending Closing. | 10 |
| 5.2 | Cooperation; Approvals, Filings and Consents. | 11 |
| 5.3 | Notice of Certain Events. | 12 |
| 5.4 | Control of Business | 12 |
| 5.5 | Mutual Cooperation | 12 |
| ARTICLE VI CONDITIONS PRECEDENT TO CLOSING | 13 | |
| 6.1 | Conditions to Obligation of Each Party | 13 |
| ARTICLE VII INDEMNIFICATION | 13 | |
| 7.1 | Indemnity By Seller. | 13 |
| 7.2 | Indemnity By Buyer. | 14 |
| 7.3 | Insurance Claims. | 14 |
| ARTICLE VIII TERMINATION, AMENDMENT, WAIVER AND EXPENSES | 14 | |
| 8.1 | Termination | 14 |
| 8.2 | Effect of Termination | 15 |
| 8.3 | Expenses | 15 |
| 8.4 | Amendment and Waiver | 15 |
| ARTICLE IX MISCELLANEOUS | 15 | |
| 9.1 | Entire Agreement | 15 |
| 9.2 | Assignment | 16 |
| 9.3 | Counterparts | 16 |
| 9.4 | Governing Law; Venue | 16 |
| 9.5 | Specific Performance | 16 |
| 9.6 | Interpretation | 16 |
| 9.7 | Severability | 17 |
| 9.8 | Notices | 17 |
| 9.9 | Representation by Counsel | 18 |
| 9.10 | Construction. | 18 |
| 9.11 | Waivers | 18 |
| 9.12 | Third Party Beneficiaries | 18 |
| 9.13 | Waiver of Jury Trial | 18 |
| SCHEDULES: | |
| Schedule 1 | Table of Definitions |
| Schedule 1.1(a) | Seller Assigned Contracts |
| Schedule 1.1(b) | Intellectual Property |
| Schedule 1.2(a) | Assumed Credits to Buyer |
| EXHIBITS: | |
| Exhibit A | Form of Trademark Assignment |
| Exhibit B | Form of Patent Assignment |
| Exhibit C | License Agreement |
| Exhibit D | Patents and Trademarks |
| ii |
ASSET PURCHASE agreement
THIS ASSET PURCHASE AGREEMENT (this “Agreement”), dated as of July 15, 2018, is made by and between, Activation Therapeutics Inc., a Delaware corporation (the “Seller”), and Olaregen Therapeutix Inc., a Delaware C Corp (the “Buyer” and/or “Olaregen”), becomes effective upon the Seller’s receipt of the total $650,000 amount due and payable pursuant to ARTICLE II, Section 2.1 paid no later than the close of business on September 16, 2018 pursuant to ARTICLE I, Section 1.3 of this Agreement.
WHEREAS, the Seller is engaged, among other things, in the business of developing and commercializing Excellagen®, an FDA cleared formulated fibrillar collagen product for use in wound care (the “Business”), and owns substantially all of the assets used in connection with the operation of the Business; and,
WHEREAS, subject to the terms and conditions set forth in this Agreement, the Seller wishes to sell, assign and transfer to the Buyer, and the Buyer wishes to purchase from the Seller, all of the Seller’s right, title and interest in and to the Acquired Assets (as defined below); and,
WHEREAS, the Seller will retain a license to exclusive rights to the Business in each of the following countries and/or territories: Greater China (including Macau), Hong Kong, Taiwan, the Russian Federation, and the Commonwealth of Independent States (“CIS”) (including Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Tajikistan, Turkmenistan and Uzbekistan) (collectively the “Retained Territories”); and
WHEREAS, the Seller will also retain a license to exclusive rights to Excellagen® in combination with the therapeutic use of exosomes (the “Licensed Technology”) and the Buyer desires to allow Seller to retain such rights; and
WHEREAS, the terms and conditions governing a license from the Buyer to the Seller of the Retained Territories and the Licensed Technology shall be duly memorialized in a license agreement between the parties, in substantially the same form as that annexed hereto as Exhibit C (the “License Agreement”); and
WHEREAS, capitalized terms used and not otherwise defined herein shall have the meanings set forth on Schedule I attached hereto.
NOW, THEREFORE, in consideration of the foregoing and the mutual representations, warranties, covenants and agreements herein set forth the Seller, and the Buyer hereby agree as follows:
| 1 |
ARTICLE I
PURCHASE AND SALE OF ASSETS
1.1 Purchase and Sale of Assets. Upon the closing of the transaction subject of this Agreement (the “Closing”) (as defined more fully below), and upon the terms and conditions herein set forth, the Seller shall sell, transfer, assign and deliver to the Buyer, and relinquish to the Buyer in perpetuity, free and clear of all Liens, all of its right, title and interest in and to the assets to be acquired (the “Acquired Assets”). For purposes of this Agreement, these Assets include the following:
(a) worldwide rights, other than those Licensed back to the Seller in the Retained Territories, to the current Excellagen® product, and the license to exclusive rights to Excellagen® in combination with the therapeutic use of exosomes;
(b) all Excellagen precursors (i.e., biological and chemical substances or materials obtained as part of the Excellagen product manufacturing process, except raw materials), formulations and potential applications;
(c) all product extensions of Excellagen including all products comprising Excellagen in combination with other biologics and small molecules, and other cellular therapies, all protocols, documents, scientific and manufacturing know-how and discoveries concerning, respecting and/or relating to the Excellagen® product, its precursors, formulations, combinations, enhancements, extensions and potential applications; all pre-clinical and clinical research and trial data for the Excellagen® product and its combinations, including any and all health, economic and patient reported outcomes data with the exception of (a) above;
(d) FDA 510(K) cleared for Excellagen®, together with any and all filing documentation;
(e) any and all other FDA applications or draft applications for Excellagen®, its precursors, combinations or derivatives;
(f) any and all applications for Excellagen®, its precursors, combinations or derivatives filed with any European regulatory body, together with any and all regulatory submissions made anywhere else in the world other than in the Retained Territories;
(g) all data received or to be received and any intellectual property filed or to be filed from the Seller’s collaboration with Orbsen Therapeutics, Ireland;
(h) all rights, title and interest in, to or under (i) each and every contract listed on Schedule 1.1(a) attached hereto; (ii) all non-competition, non-solicitation, confidentiality, assignment of invention and similar agreements to which the Seller is a party or under which the Seller possesses any actual or beneficial rights and/or interests the primary purpose of which are to provide for non-competition, non-solicitation, confidentiality, assignment of invention or similar covenants running in favor of the Seller; and, (iii) any other agreement entered into by the Seller with any third-party from the date of this Agreement through the date of the Closing, which agreement the Buyer, in its sole discretion, agrees in writing to assume. Taken together, these contracts and/or agreements shall be known as the “Assigned Contracts”;
(i) the Seller’s Intellectual Property, including patents, trademark and domain name, listed on Schedule 1.1(b); and,
(j) the Seller’s existing credit of $200,000 with Collagen Solutions Ltd.
| 2 |
1.2 Assumption of Liabilities & Reimbursement of Credit. At the Closing, and upon the terms and conditions herein set forth, the Buyer shall assume from the Seller only the following Liabilities and Credits (collectively, the “Assumed Liabilities and Credits”):
(a) obligations of the Seller, as applicable, for performance arising only after the Closing under the Assigned Contracts, to the extent that the Seller’s rights thereunder are assigned to the Buyer hereunder;
(b) a credit, in the total amount of Two Hundred Thousand Dollars ($200,000), arising from the Assigned Contracts with such amount in the form of a discount applied on future orders placed with Collagen Solutions Ltd.; and
1.3 Closing. Subject to the terms and conditions herein set forth, the Closing will take place on or before September 16, 2018 (the “Closing Date”). The Closing shall be held at the offices of Gene Biotherapeutics, 11568 Sorrento Valley Road Suite #14, San Diego CA 92121, unless another place is agreed to in writing by the parties hereto (it being understood that the Closing may be affected by the delivery of documents via e-mail, facsimile and/or overnight courier). The consummation of the transactions contemplated by this Agreement at the Closing shall be deemed to occur at 12:01 a.m. (PST) on the Closing Date.
1.4 Payment. At the Close, the Buyer shall wire or cause to be wired to the bank account of the Seller the cash amount of Four Hundred and Twenty-Five Thousand Dollars ($425,000). This is in addition to the total $225,000 deposit that has already been made to date towards this Agreement.
1.5 Transfer Documents. At the Closing, and in addition to any other Closing documents and things required by ARTICLE VI hereof, the parties shall execute and deliver, or cause to be executed and delivered, one to the other, the following documents (collectively, the “Transfer Documents”):
(a) the Seller shall execute and deliver to the Buyer one or more trademark assignments in substantially the same form as that annexed hereto as of Exhibit A (the “Trademark Assignment”), which may be necessary or required to transfer, assign and/or convey to the Buyer the trademarks, if any, being acquired by the Buyer from the Seller pursuant to this Agreement;
(b) the Seller shall execute and deliver to the Buyer one or more patent assignments in substantially the same form as that annexed hereto as Exhibit B (the “Patent Assignment”), which may be necessary or required to transfer, assign and/or convey to the Buyer the patents, if any, being acquired by the Buyer from the Seller pursuant to this Agreement; and
(c) the Seller shall execute and deliver to the Buyer the License Agreement in substantially the same form as that annexed hereto as Exhibit C (the “License Agreement”), which may be necessary or required to transfer, assign and/or convey to the Seller the licensing rights in the Retained Territories as well as the use of Excellagen in combination with exosomes pursuant to this Agreement; and
| 3 |
(d) the Seller shall execute and deliver to the Buyer all such other bills of sale, assignments, including, without limitation, intellectual property right assignments, trade name assignments and/or domain name assignments, endorsements, certificates of title, consents and other pertinent instruments and documents of conveyance and transfer in a form reasonably satisfactory to the Buyer, as the Buyer shall, in its sole discretion, deem reasonably necessary to effectuate the transfer, assignment and/or conveyance to it of the entirety of the Seller’s right, title and/or interest in and to all, or any of, the Acquired Assets.
1.6 Further Assurances. At any time and from time to time after the Closing, at the request of the Buyer and without further consideration, the Seller shall execute and deliver such other instruments of sale, transfer, conveyance, assignment and/or confirmation to the Buyer, and shall take such other and further action, as may reasonably be necessary or required to validate, ratify and/or confirm the transfer, assignment and/or conveyance of the Acquired Assets to the Buyer and the Buyer’s corresponding ownership thereof. Each of the parties hereto shall execute such other documents and take such further action as may reasonably be necessary or required to effectuate the transactions contemplated by this Agreement.
1.7 Transfer Taxes. Each Party shall bear its own tax liabilities arising from the transaction(s) contemplated by this Agreement.
ARTICLE
II
purchase price
2.1 Total Consideration. The total consideration for the Acquired Assets (the “Total Consideration”) shall be Four Million Dollars ($4,000,000) payable as follows:
(a) Six Hundred and Fifty Thousand Dollars ($650,000) in cash at the time of the Close including any previous deposits (“Total Current Consideration”).
(b) The balance of three Million Three Hundred and Fifty Thousand Dollars ($3,350,000) will be payable in quarterly installments equal to (i) 10.00% of quarterly Net Sales as defined below (“Total Additional Consideration”). Net Sales shall be defined as total sales less usual and customary sales allowances, including Hub Fees, Sales Concessions, Co-promote Fees, Cost of Goods Sold and other charges and expenses relating to the sale of comparable pharmaceutical products pursuant to Generally Accepted Accounting Practices (GAAP), assuming that the Excellagen Average Selling Price per unit (ASP) exceeds $800. In the event that the ASP is less than $800 per unit, Cost of Goods Sold shall be excluded from the computation of Net Sales.
(c) A refund to Seller of all credits (“Credits”) acquired by Buyer pursuant to section 1.2 which credit refunds will be paid to Seller by Buyer pro rata as the credits are used to purchase goods and/or services from Collagen Solutions, Ltd. Said refund of Credits will not exceed the amount of the credits described in section 1.2(b). Such Credits will be adjusted in the event that certain expenses are required to be expended in order to re-validate, re-launch and/or to re-manufacture Excellagen® with Collagen Solutions Ltd.
(d) The Buyer may seek to avoid making payments for The Total Additional Consideration and the payment for the existing Credits with existing suppliers as outlined above by making an additional lump sum payment of $2,750,000 within 120 days after the receipt of the Total Current Consideration. In the event a payment has already been made for some Credits with existing suppliers by the Buyer, then the additional lump sum payment will be adjusted accordingly.
| 4 |
(e) If the Buyer receives the funding from a major New York-based Investment Fund, as has been described to the Seller, or any other equivalent funding from a third party, the Buyer shall be obligated to pay the Seller $2,750,000 within ten (10) days of the Buyer’s receipt of such funds.
(f) In the event that the Buyer sells the Intellectual Property and the Purchase Price has not been paid in full, the Seller will be entitled to receive the unpaid balance due.
ARTICLE
III
REPRESENTATIONS AND WARRANTIES OF THE SELLER
The Seller hereby represents and warrants to the Buyer as follows:
3.1 Organization, Good Standing and Qualification of the Seller. The Seller is duly incorporated and validly existing and in corporate and tax good standing under the Laws of the State of Delaware. The Seller has all requisite power and authority, and is in possession of all Approvals necessary, to own, lease and operate the Acquired Assets and to carry on the Business as it is now being conducted.
3.2 Authorization; Binding Obligation. The Seller has all necessary power and authority to enter into this Agreement and any Related Agreements, and to transfer, deliver and/or convey the Acquired Assets to the Buyer. The execution of this Agreement and any Related Agreements, by the Seller, and consummation of the transactions subject hereof have been duly and validly authorized by all requisite action on the part of the Seller. The obligations of the Seller set forth in this Agreement and in any Related Agreement shall, at the time these agreements are executed and delivered, constitute legal, valid and binding obligations of the Seller, as applicable, enforceable against the Seller in accordance with their terms, except (i) as limited by applicable bankruptcy, insolvency, reorganization, moratorium and other Laws of general application affecting enforcement of creditors’ rights, and (ii) as limited by Laws relating to the availability of specific performance, injunctive relief or other equitable remedies.
3.3 Consents and Approvals. The execution of this Agreement and any Related Agreements, and consummation of the transactions subject hereof, are not subject to or, in any way, contingent upon, receipt, by the Seller, of any form of approval or consent from any Person, administrative entity or other Governmental Authority.
3.4 No Violation. The execution of this Agreement and any Related Agreements, and consummation of the transactions subject hereof, do not or will not (a) conflict with or violate the Organizational Documents of the Seller, (b) conflict with or violate any Law or Order applicable to the Seller or by which the Seller may be bound, or (c) result in any breach or violation of or constitute a default under, or result in the creation of a Lien on any of the Acquired Assets pursuant to, any Assigned Contract or other agreement to which the Seller is a party or is otherwise bound.
| 5 |
3.5 Legal Proceedings. There is no Action pending or, to the knowledge of the Seller, threatened against or affecting the Seller that would (a) give any Person the right to enjoin or rescind the transactions subject of this Agreement or (b) otherwise prevent the Seller from executing and delivering this Agreement and any Related Agreements to the Buyer and/or from fulfilling its obligations to the Buyer hereunder or thereunder.
3.6 Title to Assets.
(a) The Seller is the sole and exclusive legal and beneficial owner of all right, title and interest in, and has good, valid and marketable title to, all of the Acquired Assets. The Seller has the power, authority and right to transfer, sell, assign and/or convey, to the Buyer good, valid and marketable title to all of the Acquired Assets.
(b) The Seller is not in breach of or in material default under the Assigned Contracts, or any of them; nor does there exist any event, condition or occurrence which (with or without due notice or lapse of time, or both) would give rise to or constitute such a breach or default on the part of the Seller. To the knowledge of the Seller, no counter-party to the Assigned Contracts, or any of them, is in default under the terms thereof; nor, to the knowledge of the Seller, does there exist any event, condition or occurrence which (with or without due notice or lapse of time, or both) would give rise to or constitute such a breach or default on the part of any such counter-party.
(c) Unless otherwise agreed, at the Closing, the Assigned Contracts, and each of them, shall be in full force and effect, and the obligations therein set forth shall be valid and binding upon the Seller, on the one hand, and any designated counter-party, on the other, provided, however, that the executor obligations of the counter-party may be contingent on the receipt of a balance owed, as set forth in Schedule 1.2(a). Other than stated balances owed, the Seller has no knowledge of any claim, dispute or controversy with respect to the Assigned Contracts, or any of them. For purposes of clarification, the Seller is responsible for any and all liabilities with said counter-parties prior to the Closing.
(d) The execution of this Agreement and consummation of the transactions subject hereof will not conflict with, or have any material impact upon, the terms and/or conditions of the Assigned Contracts, or any of them.
(e) In the event that the Assets are leveraged by the Buyer, and the Purchase Price has not been fully paid to the Seller, the Buyer, or successor to the Buyer, agrees that any creditor will need to write appropriate language that establishes the priority of the remaining unpaid balance of the Purchase Price due to the Seller.
(i) In the case of a liquidation event by the Buyer, or a successor of the Buyer, and the Assets must be liquidated, either voluntary or involuntary, then the proceeds from the sale of the Assets shall have priority to paydown the unpaid Purchase Price due to the Seller (the “Liquidation Preference”).
(ii) In the event that the amount of the Liquidation Preference is not sufficient to cover the unpaid Purchase Price due to the Seller, then the Seller shall be entitled to a return of the Intellectual Property.
| 6 |
3.7 Intellectual Property.
(a) Schedule 1.1(b) sets forth a complete and accurate list of all United States and foreign patents, trademarks (including unregistered trademarks), internet domain names and other items comprising the Seller’s Intellectual Property, indicating for each, the applicable jurisdiction, registration number (or application number), date issued (or date filed), and status (including the next action or payment and date due). To the knowledge of the Seller, there has been no prior use of any such trademark(s) by any Person which would confer upon such Person superior rights in or to such trademark(s). No patent included in the Seller’s Intellectual Property has been or is now involved in any litigation, infringement, interference, reissue, re-examination, opposition, invalidity or nullity proceeding. To the knowledge of the Seller, there are no trademarks owned by any third- party which potentially conflict with the trademark(s) included in the Seller’s Intellectual Property. Nor, to the knowledge of the Seller, are there any patents owned by any third-party which potentially interfere with the patents included in the Seller’s Intellectual Property, other than art cited by the examiner in the course of patent examination.
(b) The Seller’s Intellectual Property listed on Schedule 1.1(b) constitutes all of the Intellectual Property used in connection with or necessary for the conduct of the Business as currently conducted, including all Intellectual Property necessary to use, manufacture, market and distribute the Excellagen® product.
(c) The Seller is the sole and exclusive owner of all of the Seller’s Intellectual Property listed on Schedule 1.1(b) and has a lawful, valid, and enforceable right to transfer said Intellectual Property to the Buyer free and clear of any and all Liens. All of the Seller’s Intellectual Property has been created by employees of the Seller, acting within the scope of their employment, or by independent contractors of the Seller, who have all executed agreements expressly assigning all right, title and interest in or to such Intellectual Property to the Seller. No portion of the Seller’s Intellectual Property was jointly developed with any independent third party.
3.8 FDA Compliance; Compliance with Healthcare Laws.
(a) The operations of the Business and, to the knowledge of the Seller, those of any third party involved in the Business, including the manufacture, import, export, testing, development, processing, packaging, labeling, storage, marketing, and distribution of the Excellagen® product, are in compliance, in all material respects, with all applicable Laws, Approvals and Orders administered or issued by the FDA or any foreign regulatory agency with a similar regulatory purpose. There are no pending or, to the knowledge of the Seller, threatened Actions by the FDA or any other similar foreign regulatory agency against the Seller arising out of or relating to the Business and/or its operations. The Seller has not received notice of any pending or threatened claim related to a violation of any Law within the FDA’s regulatory jurisdiction (or any similar foreign law, rule regulation or policy); nor does the Seller have any knowledge or reason to believe that the FDA or any similar foreign regulatory agency is considering such action.
| 7 |
(b) The Seller has not received any Form 483 notice of adverse findings, warning letter, correspondence or notice from the FDA, or similar foreign regulatory agency, alleging or asserting noncompliance, by the Seller, with any applicable Laws enforced by the FDA or such similar foreign regulatory agency. Nor does the Seller have any knowledge or reason to believe that any such notice, warning or correspondence from either the FDA or any similar foreign regulatory agency will be forthcoming.
(c) At all times material, the manufacture of Excellagen® by or on behalf of the Seller has been conducted in full compliance with all applicable Laws and Regulations, including the FDA’s Quality Systems Regulation. In addition, the Seller, and to the knowledge of the Seller, any manufacturer of Excellagen® on the Seller’s behalf, is and, at all times material has been, in compliance with all applicable FDA requirements, including, without limitation, the registration, listing and premarket notification requirements set forth in 21 U.S.C. § 360, the investigational device exemption set forth in 21 U.S.C. § 360j(g), as well as any similar requirements applicable in any foreign jurisdiction.
(d) The Seller is not subject to any determination by a Governmental Authority excluding, suspending, debarring or otherwise restricting, or proposing to restrict, the Seller, or any director, officer, employee, contractor, or agent thereof, from participation in any government health care program, whether pursuant to 42 U.S.C. § 1320a-7(a) or otherwise.
(e) The Seller is not the subject of any pending or, to the knowledge of the Seller, threatened, investigation by the FDA pursuant to its “Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities,” Final Policy set forth in 56 Fed. Reg. 46191 (September 10, 1991), or any similar investigation by any foreign regulatory agency. The Seller has not committed any act, made any statement, or failed to make any statement that would provide a basis for the FDA to act adversely against the Seller under the foregoing Final Policy, and has not committed any act, made any statement or failed to make any statement that would provide a basis for any foreign regulatory agency to act adversely against the Seller under a similar policy. The Seller has not employed, in any capacity, any individual who has been debarred or excluded pursuant to the Food, Drug, & Cosmetics Act or 42 U.S.C. § 1320a-7(a), nor has the Seller used, employed, hired or contracted with any clinical investigator who has been disqualified under 21 C.F.R. § 812.119 or who has engaged in any conduct that would reasonably be expected to result in disqualification as a clinical investigator under 21 C.F.R. § 812.119. Similarly, the Seller has not used, in connection with any clinical investigation conducted by or on its behalf, any institutional review board or institution that has been disqualified under 21 C.F.R. § 56.121 or that has engaged in conduct that would reasonably be expected to result in disqualification under 21 C.F.R. § 56.121.
(f) Any clinical trials or human and animal studies pertaining to the Business and/or the Excellagen® product were and, if still pending, are being conducted (to the Seller’s knowledge with respect to such studies conducted by third parties) in all material respects in accordance with standard medical and scientific research procedures and all applicable rules, regulations and policies of the FDA, including its current Good Clinical Practices and Good Laboratory Practices, and in compliance with all applicable domestic and foreign regulatory requirements and/or standards.
(g) The Seller has not knowingly or willfully solicited, received, paid or offered to pay any remuneration, directly or indirectly, overtly or covertly, in cash or kind, for the purpose of making or receiving any referral which violated or may have violated any applicable state or federal anti-kickback Law, including without limitation the Federal Health Care Program Anti-Kickback Statute, 42 U.S.C. § 1320a-7b(b) (known as the “Anti-Kickback Statute”).
| 8 |
(h) The Seller recognizes that in the event of a liquidation event by the Buyer where all the assets of Buyer including the Business Assets of the Seller that were acquired with this Asset Purchase Agreement must be liquidated, then the proceeds from the sale of these combined assets will first be used to pay the outstanding and remaining balance due to meet the Total Additional Consideration due to the Seller. The Seller further recognizes that in the event that this liquidation amount is not sufficient to cover the remaining and outstanding Total Consideration then the Seller shall have no further recourse and/or claim against the Buyer.
ARTICLE
IV
REPRESENTATIONS AND WARRANTIES OF THE BUYER
The Buyer hereby represents and warrants to the Seller as follows:
4.1 Organization and Good Standing. The Buyer is a limited liability company duly organized, validly existing and in good standing under the Laws of the State of Delaware.
4.2 Authorization; Binding Obligation. The Buyer has all necessary power and authority to enter into this Agreement and any Related Agreements and to consummate the transactions subject hereof and thereof. The execution of this Agreement and any Related Agreements, by the Buyer, and the consummation of the transactions subject hereof have been duly and validly authorized by all requisite action on the part of the Buyer. The obligations of the Buyer set forth in this Agreement and in any Related Agreement shall, at the time these agreements are executed and delivered, constitute legal, valid, and binding obligations of the Buyer, as applicable, and shall be enforceable against the Buyer in accordance with their terms, except (i) as limited by applicable bankruptcy, insolvency, reorganization, moratorium and other Laws of general application affecting enforcement of creditors’ rights, and (ii) as limited by Laws relating to the availability of specific performance, injunctive relief or other equitable remedies.
4.3 Consents and Approvals. The execution of this Agreement and any Related Agreements, and consummation of the transactions subject hereof, are not subject to or, in any way, contingent upon, receipt, by the Buyer, of any form of approval or consent from any person, administrative entity or other Governmental Authority.
4.4 No Violation. The execution of this Agreement and any Related Agreements, and consummation of the transactions subject hereof, do not or will not (a) conflict with or violate the Organizational Documents of the Buyer, (b) conflict with or violate any Law or Order applicable to the Buyer or by which the Buyer may be bound, or (c) result in any breach or violation of, or constitute a default under, any agreement to which the Buyer is a party, except where such conflict or breach would not reasonably be likely to have a material adverse impact upon the Buyer’s ability to consummate the transactions subject of this Agreement.
4.5 Legal Proceedings. There is no Action pending or, to the knowledge of the Buyer, threatened, against or affecting the Buyer that would (a) give any Person the right to enjoin or rescind the transactions subject of this Agreement or (b) otherwise prevent the Buyer from executing and delivering this Agreement and any Related Agreements to the Seller and/or from fulfilling its obligations to the Seller hereunder or thereunder.
| 9 |
4.6 Audit Rights. the Buyer recognizes that the Seller will be entitled to inspect the sales records of Olaregen with four (4) weeks prior written notice not more than once a year during business hours, by an independent auditor, which is under a professional duty of confidentiality elected by the Seller. The cost of such inspection shall be borne by the Seller. These Audit Rights will continue until the total consideration has been paid in full or once the Option to avoid making further payments has been exercised as described above in Article II (d).
4.7 Seniority of the Total Additional Consideration. In the event the Business Assets are levered by the Buyer, and the Total Additional Consideration has not been fully paid to the Seller, Olaregen agrees that any lender will need to write appropriate language that establishes the priority of the remaining balance due on the Total Additional Consideration to the Seller.
4.8 Accelerated Payment of Total Additional Consideration. In the event the Buyer sells the Intellectual Property, and the Total Additional Consideration has not been paid in full, the Seller will be entitled to receive the balance due and remaining on the Total Additional Consideration prior to any other payments being made.
4.9 Financial Ability. The Buyer has earmarked sufficient funds available to consummate the transactions subject of this Agreement and to fulfill its obligations to the Seller pursuant to this Agreement and any Related Agreements.
ARTICLE
V
COVENANTS
5.1 Conduct of Business Pending Closing.
(a) The Seller covenants and agrees that, from the date this Agreement is executed through the Closing, or upon the earlier termination of this Agreement, it shall:
(i) preserve intact all of the Acquired Assets in the ordinary course of Business and in a manner consistent with past practice; and,
(ii) use commercially reasonable efforts to enter into new agreements with each and every counter-party to the Assigned Contracts, both active and expired.
(b) The Seller covenants and agrees that, from the date this Agreement is executed through the Closing, or upon the earlier termination of this Agreement, -it shall not:
(i) sell, transfer, convey, assign, lease, license, pledge or otherwise dispose of any of the Acquired Assets, or any of them;
| 10 |
(ii) collateralize, encumber, impair or cause any Lien to be imposed upon the Acquired Assets, or any of them;
(iii) amend or modify in any material way any existing agreements respecting, concerning or pertaining to the Acquired Assets, or any of them;
(iv) discharge, compromise, satisfy, resolve or commence any Action or waive, assign or release any material rights or claims relating to the Business and/or the Acquired Assets;
(v) engage in, adopt a plan of and/or begin a complete or partial liquidation, dissolution, merger, consolidation, restructuring, recapitalization or other reorganization; or,
(vi) take any action or fail to take any action that would result in any of the representations and warranties set forth in ARTICLE III being rendered false or inaccurate, or which, individually or in the aggregate, would have a have a Business Material Adverse Effect or otherwise materially impair the ability of the Seller to consummate the transactions subject of this Agreement.
5.2 Cooperation; Approvals, Filings and Consents.
(a) Upon the terms and subject to the conditions set forth in this Agreement, each of the parties hereto shall use commercially reasonable efforts to take, or cause to be taken, all actions, and do, or cause to be done, and to assist and cooperate with the other in doing, all things necessary, proper or advisable to consummate the transactions subject of this Agreement and to satisfy or cause to be satisfied all of the conditions precedent set forth in ARTICLE VI, as applicable to each of them.
(b) The Seller, and the Buyer shall, as promptly as practicable, take any and all commercially reasonable steps necessary to obtain all required Approvals, if any, from Governmental Authorities and to make all other necessary registrations and filings, if any, which may be required to execute and deliver this Agreement and any Related Agreements and to consummate the transactions subject hereof and thereof.
(c) The Seller shall, as promptly as practicable, use its best efforts to obtain all other consents from third parties that, in the reasonable discretion of the Buyer, are necessary or desirable to consummate the transactions subject of this Agreement (“Third Party Consents”).
(d) The Seller shall make commercially reasonable efforts to arrange for the drafting and filing of additional patent applications relating to the Excellagen® product and/or the Business in the European Union, as well as in Asia, the Middle East, and Latin America (the “New Patents”). The cost of the New Patents will be borne by Buyer, with the exception of the PCT patent application which shall be borne by the Seller, including patent attorney fees and filing costs; provided, however, the Buyer will not be obligated to reimburse the Seller for time spent arranging the drafting and filing of the New Patents.
| 11 |
5.3 Notice of Certain Events.
(a) During the period from the date this Agreement is executed through the Closing, or upon the earlier termination of this Agreement, the Seller shall promptly notify the Buyer in writing of: (i) any event, condition, fact or circumstance that occurred or existed on or prior to the effective date of this Agreement, respecting which the Seller becomes aware following the execution of this Agreement, which has caused or constitutes an inaccuracy in and/or breach of any representation or warranty made by the Seller herein; (ii) any event, condition, fact or circumstance that occurs, arises or exists after the effective date of this Agreement, respecting which the Seller becomes aware, which has caused or constitutes an inaccuracy in or breach of any representation or warranty made by the Seller herein and, (iii) any material breach of any covenant or obligation herein set forth on the part of the Seller.
(b) In addition to the foregoing, during the period from the date this Agreement is executed through the Closing, or upon the earlier termination of this Agreement, the Seller shall give prompt written notice to the Buyer of (i) any notice or other communication received from any Person alleging that the consent of such Person is or may be required to consummate the transactions subject of this Agreement, or any of them, (ii) any notice or other communication received from any Governmental Authority concerning, pertaining to or respecting any Approval which may be necessary or required to consummate the transactions subject of this Agreement, or any of them, (iii) any Action commenced or threatened concerning, involving or relating to the Business, the Acquired Assets, the Seller’s Intellectual Property and/or the transactions subject of this Agreement, (iv) the occurrence of any event of breach or default under, the Assigned Contracts, or any of them, (v) any notice or other communication received from any Person concerning, involving or respecting the Assigned Contracts, or any of them, and, (vi) any change, event or circumstance which would reasonably be expected to materially delay or impede the ability of the Seller to consummate the transactions subject of this Agreement and/or to fulfill its contractual obligations to the Buyer hereunder.
(c) During the period from the date this Agreement is executed through the Closing, or upon the earlier termination of this Agreement, the Buyer shall give prompt written notice to the Seller of (i) any notice or other communication received from any Person alleging that the consent of such Person is or may be required to consummate the transactions subject of this Agreement, or any of them, (ii) any notice or other communication received from any Governmental Authority concerning, pertaining to or respecting the transactions subject of this Agreement, or any of them, or, (iii) any change, event or circumstance which would reasonably be expected to materially delay or impede the ability of the Buyer to consummate the transactions subject of this agreement and/or to fulfill its contractual obligations to the Seller hereunder.
5.4 Control of Business. Nothing contained in this Agreement shall give the Buyer the right to control or direct the operations of the Business, directly or indirectly, prior to the Closing. Prior to the Closing, the Seller shall exercise complete control and supervision over the operations of the Business, consistent with the terms and conditions herein set forth.
5.5 Mutual Cooperation. From and after the Closing Date, the Seller, on the one hand, and the Buyer, on the other, shall use their respective best and reasonable efforts to provide the other with such books, records and information, and to make available to the other such personnel or representatives, as reasonably may be necessary to enable either party (i) to respond to pertinent inquiries received from any Governmental Authority or third-party professional, (ii) to prepare any applicable filings (including tax filings), or (iii) to prosecute or defend against any Action concerning, arising out of and/or relating to the conduct of the Business prior to or after the Closing Date. It is understood and agreed that the party who seeks and receives from the other any form of the assistance provided for above shall promptly reimburse the latter for any and all out-of-pocket expenses incurred and paid in connection with the rendition of such assistance, including reasonable legal fees and disbursements. Neither party shall be obligated to reimburse the other for the value of any time spent rendering the assistance provided for in this paragraph, irrespective of the status (e.g., employee, independent contractor, consultant) of the Person(s) rendering any such assistance.
| 12 |
ARTICLE
VI
CONDITIONS PRECEDENT TO CLOSING
6.1 Conditions to Obligation of Each Party. The respective obligations of the parties to close upon the transactions subject of this Agreement are contingent upon the following:
(a) Governmental Approvals. All necessary Approvals from Governmental Authorities, if any, shall have been obtained, and any applicable waiting periods shall have expired.
(b) No Injunctions or Restraints; Illegality. No temporary restraining order, preliminary or permanent injunction or other Order (whether temporary, preliminary or permanent) issued by any Court of competent jurisdiction or other legal restraint or prohibition shall have issued barring, preventing or interfering, in any way, with the consummation of the transactions subject of this Agreement, or any of them; nor shall any Action seeking any form of such relief then be pending.
(c) Representations and Warranties. The representations and warranties of both the Seller and the Buyer, as set forth, respectively, in Articles III and IV of this Agreement shall be true and correct in all material respects, both at the time this Agreement is executed and as of the Closing Date (except for representations and warranties made as of a specified date, the legitimacy and accuracy of which will be determined only as of that date).
(d) Purchase Price. The Buyer shall have tendered to the Seller the consideration provided for in Section 2.1(a) above.
ARTICLE
VII
INDEMNIFICATION
7.1 Indemnity By Seller. The Seller shall indemnify and hold harmless the Buyer, its members, officers, directors, employees, agents, affiliates and customers from and against any and all costs (including reasonable legal fees), damages, expenses, losses, suits, claims and demands, in any manner caused by, resulting from or arising out of (i) any breach of this Agreement by the Seller or, (ii) any third-party claim or suit arising out of or relating to the negligence or willful misconduct on the part of the Seller, its affiliates, officers, directors, employees, agents and/or representatives.
| 13 |
7.2 Indemnity By Buyer. The Buyer shall indemnify and hold harmless the Seller, its affiliates, officers, directors, employees, agents and customers from and against any and all costs (including reasonable legal fees), damages, expenses, losses, suits, claims and demands, in any manner caused by, resulting from or arising out of (i) any breach of this Agreement by the Buyer or, (ii) any third-party claim or suit arising out of or relating to the negligence or willful misconduct on the part of the Buyer, its members, affiliates, officers, directors, employees, agents and/or representatives.
7.3 Insurance Claims. If Losses with respect to any claim for indemnification pursuant to Section 7.1 may be subject to coverage under any policy of insurance owned by the Seller, the Seller shall, at the request of the Buyer, and without further consideration, use commercially reasonable efforts to seek and recover the value of such Losses from the carrier who issued said policy and, thereafter, pay over any sums recovered to the Buyer. If Losses with respect to any claim for indemnification pursuant to Section 7.2 may be subject to coverage under any policy of insurance owned by the Buyer, the Buyer shall, at the request of the Seller, and without further consideration, use commercially reasonable efforts to seek and recover the value of such Losses from the carrier who issued said policy and, thereafter, pay over any sums recovered to the Seller.
ARTICLE
VIII
TERMINATION, AMENDMENT, WAIVER AND EXPENSES
8.1 Termination. Following its execution, this Agreement may be terminated, and the transactions subject hereof cancelled or abandoned at any time prior to the Closing upon the occurrence of one or more of the following conditions:
(a) By written mutual consent of both the Buyer and the Seller;
(b) By either the Buyer or the Seller if the Closing shall not have occurred on or before September 16, 2018 (the “Outside Date”); provided, however, that the right to terminate this Agreement under this sub-section shall not be available to any party whose failure to satisfy or fulfill its obligations hereunder results in, or is the cause of, the failure to close on or before such date;
(c) By either the Buyer or the Seller if any Court or Governmental Authority shall have issued a final and non-appealable Order or similar mandate restraining, enjoining or otherwise prohibiting the Closing;
(d) By the Buyer, if the Buyer is not in material breach of any its obligations under this Agreement, and the Seller is in material breach of its representations, warranties or obligations hereunder, which breach is incapable of being cured or, if capable of being cured, is not cured prior to the earlier of (a) the Business Day before the Outside Date, or (b) within twenty (20) calendar days of the date on which the Seller is notified by the Buyer of any such material breach; or
| 14 |
(e) By the Seller, if the Seller is not in material breach of any of its obligations under this Agreement, and the Buyer is in material breach of its representations, warranties or obligations hereunder, which breach is incapable of being cured or, if capable of being cured is not cured prior to the earlier of (i) the Business Day before the Outside Date, or (ii) within twenty (20) days of the date on which the Buyer is notified by the Seller of any such material breach.
8.2 Effect of Termination. In the event this Agreement is terminated pursuant to Section 8.1 hereof, it shall become null and void, and the parties respective obligations, one to the other, as herein set forth, shall cease; provided, however, that if, at the time of termination, one party has sustained Losses arising out of or resulting from any prior material breach of this Agreement by the other, said party shall retain the right to seek recompense from the other in respect of any and all Losses so sustained.
8.3 Expenses. Each of the parties hereto shall bear responsibility for payment of any and all fees, costs and expenses incurred by it in connection with the negotiation and preparation of this Agreement and/or in furtherance the transactions contemplated hereby, including, without limitation, legal, accounting and consulting fees, whether or not the Closing occurs. The foregoing notwithstanding, the parties hereto may agree, at any time prior to the Closing, to share in certain costs and/or expenses associated with the completion of the transactions subject of this Agreement.
8.4 Amendment and Waiver. This Agreement may only be amended in writing, by an instrument duly authorized and executed by the Buyer and the Seller and/or their respective representatives. Each of the parties hereto may seek to extend the time within which to perform or satisfy its respective obligations to the other pursuant to this Agreement, or, in lieu thereof, to waive the performance of such obligations, or any of them. Any such extension or waiver sought shall be valid only if set forth in a writing duly authorized and executed by both the Buyer and the Seller and/or their respective representatives.
ARTICLE
IX
MISCELLANEOUS
9.1 Entire Agreement. This Agreement contains the entire understanding of the parties with respect to the subject matter hereof and the transactions contemplated hereby, and all prior negotiations, agreements and undertakings, both written and oral, with respect hereto are merged into this Agreement. Each party hereto expressly acknowledges that, in executing and delivering this Agreement, it is not relying upon any warranties, representations, promises or statements, except to the extent the same are expressly set forth in this Agreement. The Related Agreements, together with the Schedules and Exhibits annexed hereto and any other agreements, documents and instruments to be delivered in connection herewith, are hereby incorporated by reference and made a part of this Agreement. (See also, Section 9.6 below.) The foregoing notwithstanding, it is understood and agreed that the Confidentiality Agreement shall survive the execution of this Agreement pending consummation of the transactions subject hereof, following which it shall terminate.
| 15 |
9.2 Assignment. The Seller shall not assign or transfer this Agreement or any of its rights hereunder, or delegate any of its obligations hereunder to any third-party, without the prior written consent of the Buyer, unless such assignment is being made to an entity which is an Affiliate of the Seller (as defined in Schedule I hereto), in which case no specific consent will be required. The Buyer may assign or otherwise transfer this Agreement or any of its rights hereunder, or delegate any of its obligations hereunder; provided, however, that no such assignment or transfer shall relieve the Buyer of its obligations to the Seller pursuant to this Agreement. Subject to the foregoing, this Agreement and the rights and obligations herein set forth shall inure to the benefit of, and be binding upon, the parties hereto, and each of their respective successors, heirs and permitted assigns.
9.3 Counterparts. This Agreement may be executed in counterparts, each of which shall be deemed an original, and all of which together shall be deemed one and the same instrument. For the convenience of the parties, faxed and/or electronic signatures shall be treated as, and deemed to be, original signatures.
9.4 Governing Law; Venue. This Agreement shall be governed by the Laws of the State of New York without giving effect to any choice of law or conflict of law provision or rule. All Actions arising out of or relating to this Agreement shall be heard and determined in any state or federal court sitting in the State of New York, County of New York. Each of the parties to this Agreement irrevocably submits to the exclusive jurisdiction of the state courts of New York and to the jurisdiction of the United States District Court for the Southern District of New York for the purpose of any Action arising out of or relating to this Agreement. Each of the parties to this Agreement consents to service of process by delivery in accordance with the provisions set forth in Section 9.8 hereof.
9.5 Specific Performance. The rights and remedies of the parties hereto shall be cumulative. The transactions contemplated by this Agreement are unique transactions and any failure on the part of either of the parties hereto to consummate these transactions on the terms and conditions herein set forth may not be fully compensable in damages. Any actual or threatened breach of this Agreement by either of the parties hereto, therefore, may cause the other to suffer irreparable harm. Accordingly, and in addition to any other remedies which may be available to either of the parties hereto arising out of any actual or threatened breach of this Agreement by the other, the non-breaching party shall also have the right to seek specific performance of this Agreement.
9.6 Interpretation. The Schedules and Exhibits annexed hereto are an integral part of this Agreement, and each of them is incorporated herein by reference. As such, unless otherwise indicated, all references herein to this “Agreement” shall be deemed to encompass or include both this Agreement and the annexed Schedules and Exhibits. Other than for words which start a sentence or paragraph, capitalized terms in this Agreement, its recitals, Schedules and Exhibits, shall have the meaning ascribed to them in Schedule I hereto. Words not so defined in this Agreement shall have the meaning found in Merriam-Webster’s Dictionary. In addition, when used herein, the words “include,” “includes” and “including” shall be deemed, in each case, to be followed by the words “without limitation.” Similarly, unless otherwise indicated, the word “herein” and similar references shall be deemed to encompass or include the entire Agreement, rather than any specific provision or section hereof. The Table of Contents and the headings contained in this Agreement are for reference purposes only and shall not, in any way, affect the meaning or substance of this Agreement. The term “$” shall mean United States dollars. For purposes of construction, all pronouns herein set forth shall be deemed to include or encompass the masculine, feminine, neuter, singular and plural forms thereof. Finally, any reference to the Buyer or Seller found in this Agreement shall be deemed to encompass or include each party, together with their respective Affiliates.
| 16 |
9.7 Severability. If any term or provision of this Agreement is found to be invalid, illegal or incapable of being performed, all other terms and provisions of this Agreement shall nevertheless remain in full force and be valid and enforceable to the fullest extent permitted by law; provided, however, that in the event any such finding or determination is made, the parties hereto agree to use their best efforts to modify, supplement and/or amend this Agreement, as may reasonably be necessary, to enable them to consummate the transactions subject of this Agreement, if possible.
9.8 Notices. All notices or other communications which are required or permitted hereunder shall be in writing and shall be sufficient if delivered personally, by electronic mail, by overnight courier or by registered or certified mail, postage prepaid, return receipt requested, to the following:
| If to the Buyer: | |
| Olaregen Therapeutix Inc. | |
| Mr. Anthony Dolisi | |
| Chief Executive Officer | |
| 1001 Avenue of the Americas, 10th floor | |
| New York, NY 10018 | |
| Email: tonydolisi@yahoo.com | |
| with a copy to (which shall not constitute notice): | |
| Stuart L. Melnick, Esq. | |
| Law Offices Stuart L. Melnick, LLC | |
| 315 Madison Avenue, Suite 901 | |
| New York, NY 10017 | |
| Email: stuart@melnick-law.com | |
| If to the Seller: | |
| Gene Biotherapeutics Inc. | |
| Chris Reinhard, Chief Executive Officer | |
| 11568 Sorrento Valley Rd., Suite 14 | |
| San Diego, CA 92121 | |
| creinhard@angionetics.com |
| 17 |
| with a copy to (which shall not constitute notice): | |
| James A. Mercer III | |
| Sheppard Mullin Richter & Hampton | |
| 12275 El Camino Real, Suite 200 | |
| San Diego, CA 92130 | |
| jmercer@sheppardmullin.com |
or to such other address as the party to whom notice is to be given may have furnished to the other in writing. All such notices or communications shall be deemed to be received (a) in the case of personal delivery, on the date of such delivery, (b) in the case of overnight courier, on the next Business Day after the date when sent, (c) in the case of electronic mail, on the date sent, and, (d) in the case of regular mail, on the second Business Day following the date on which the mailing containing such communication was posted for delivery.
9.9 Representation by Counsel. Each of the parties hereto acknowledges that it has been advised by legal counsel in the course of negotiations leading to the formation and execution of this Agreement and any Related Agreements. Each also acknowledges that it has had a full and fair opportunity to review and inquire about this Agreement, the Related Agreements, the Assigned Contracts, applicable Schedules and Exhibits, and other pertinent documentation, as well as the substance and construction thereof.
9.10 Construction.The parties have participated jointly in the negotiations leading to the formation of this Agreement; each also has participated in the drafting of this Agreement. As such, in the event any ambiguity or question of intent or interpretation arises, there shall be no presumption or burden of proof favoring or disfavoring either of the parties hereto.
9.11 Waivers. The failure on the part of any party to this Agreement to take action against the other arising out of or by reason of the latter’s breach of any term or provision of this Agreement shall not constitute a waiver of that party’s right to enforce any other term or provision of this Agreement or to take action against such breach or default or any subsequent breach or default. To be effective any waiver must be in writing and signed by the waiving party.
9.12 Third Party Beneficiaries. Nothing in this Agreement is intended to confer, nor may any term or provision set forth in this Agreement be construed to confer, any rights, interests, remedies, obligations or liabilities upon any Person other than the parties hereto, their respective representatives, successors and/or assigns. The representations and warranties contained in this Agreement are for the sole benefit of the parties hereto and are not to be relied upon by any other Person for any reason or purpose whatsoever.
9.13 Waiver of Jury Trial. THE PARTIES HERETO ACKNOWLEDGE AND AGREE THAT ANY CONTROVERSY WHICH MAY ARISE UNDER THIS AGREEMENT IS LIKELY TO INVOLVE COMPLICATED AND DIFFICULT ISSUES. EACH, THEREFORE, IRREVOCABLY AND UNCONDITIONALLY WAIVES ANY RIGHT IT MAY HAVE TO A TRIAL BY JURY IN CONNECTION WITH ANY LITIGATION DIRECTLY OR INDIRECTLY ARISING OUT OF OR RELATING TO THIS AGREEMENT OR THE TRANSACTIONS CONTEMPLATED HEREBY.
| 18 |
NOW, THEREFORE, the parties hereto have executed or caused this Asset Purchase Agreement to be executed, by their duly authorized representatives, as of the date first written above.
| BUYER: | ||
| OLAREGEN THERAPEUTIX INC. | ||
| By: | /s/ Anthony Dolisi | |
| Name: | Anthony Dolisi | |
| Title: | Chief Executive Officer | |
| SELLER: | ||
| ACTIVATION THERAPEUTICS INC. | ||
| By: | /s/ Christopher J. Reinhard | |
| Name: | Christopher J. Reinhard | |
| Title: | Chief Executive Officer | |
EXHIBIT 21
SUBSIDIARIES
| Name of Subsidiary | Jurisdiction of Organization | |
| Activation Therapeutics, Inc. | Delaware | |
| Angionetics, Inc. | Delaware |
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Christopher J. Reinhard, certify that:
1.
I have reviewed this report on Form 10-K of
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4.
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and
procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined
in Exchange Act Rules 13a-15(f) and 15d-15(
| a. | designed
such disclosure controls and procedures, or caused such disclosure controls and procedures
to be designed under our supervision, to ensure that material information relating to
the registrant, including its consolidated subsidiaries, is made known to us by others
within those entities, particularly during the period in which this report is being prepared; | |
| b. | designed
such internal control over financial reporting, or caused such internal control over
financial reporting to be designed under our supervision, to provide reasonable assurance
regarding the reliability of financial reporting and the preparation of financial statements
for external purposes in accordance with generally accepted accounting principles; | |
| c. | evaluated
the effectiveness of the registrant | |
| d. | disclosed
in this report any change in the registrant |
5.
The registrant
| a. | all
significant deficiencies and material weaknesses in the design or operation of internal
control over financial reporting which are reasonably likely to adversely affect the
registrant | |
| b. | any
fraud, whether or not material, that involves management or other employees who have
a significant role in the registrant |
|
/s/ Christopher J. Reinhard |
| Christopher
J. | |
| Chief Executive Officer |
EXHIBIT 31.2
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350
AS ADOPTED PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, James L. Grainer, certify that:
1. I have reviewed this report on Form 10-K of Gene Biotherapeutics, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
| a. | designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; | |
| b. | designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; | |
| c. | evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and | |
| d. | disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
5. The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
| a. | all significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and | |
| b. | any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Dated: April 23, 2021
| /s/ James L. Grainer | |
| James L. Grainer | |
| Chief Financial Officer |
EXHIBIT 32.1
CERTIFICATION OF CHIEF EXECUTIVE OFFICER AND CHIEF FINANCIAL OFFICER PURSUANT TO
18 U.S.C. SECTION 1350
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
I, Christopher J. Reinhard, certify, as of the date hereof, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that the Annual Report of Gene Biotherapeutics, Inc. on Form 10-K for the fiscal year ended December 31, 2019 with expanded financial and other disclosures in lieu of filing a separate Annual Report on Form 10-K for the fiscal years ended December 31, 2018 and December 31, 2017, fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, and that the information contained in the Form 10-K fairly presents, in all material respects, the financial condition and results of operations of Gene Biotherapeutics, Inc.as of, and for the periods presented.
| Date: April 23, 2021 | |
| /s/ Christopher J. Reinhard | |
| Christopher J. Reinhard | |
| Chief Executive Officer |
I, James L. Grainer, certify, as of the date hereof, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, that the Annual Report of Gene Biotherapeutics, Inc. on Form 10-K for the fiscal year ended December 31, 2019 with expanded financial and other disclosures in lieu of filing a separate Annual Report on Form 10-K for the fiscal years ended December 31, 2018 and December 31, 2017, fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, and that the information contained in the Form 10-K fairly presents, in all material respects, the financial condition and results of operations of Gene Biotherapeutics, Inc.as of, and for the periods presented.
| Date: April 23, 2021 | |
| /s/ James L. Grainer | |
| James L. Grainer | |
| Chief Financial Officer |
A signed original of this written statement required by Section 906 has been provided to the Company and will be furnished to the Securities and Exchange Commission or its staff upon request.